Carbon-13 NMR
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Structure Determination of Organic Compounds
While proton magnetic resonance (PMR) is the most common type of NMR, it is also possible to observe other nuclei which have spin quantum numbers not equal to zero.
CARBON-13 NMR
While
proton magnetic resonance (PMR) is the most common type of NMR, it is also
possible to observe other nuclei which have spin quantum numbers not equal to
zero. Of greatest interest to organic chemists is 13C NMR
spectroscopy. Carbon-13 has a spin quantum number I = 1/2 , the same as a
proton, so that when placed in a magnetic field, two possible orientations with
respect to the field are possible — one of lower energy and one of higher
energy. Transitions between these two spin states occur at discrete frequencies
in the radio frequency region. Absorption of energy at the resonance frequency
causes nuclei in the lower energy level (aligned) to undergo a transition to
the higher energy level (opposed). This process is the same as discussed
previously for protons, and the equations which govern the absorption are the
same and will not be repeated.
There
are significant differences between a 13C nucleus and a proton which
must be dealt with:
1.
Low (∼1%) natural abundance of 13C.
2. Lower magnetogyric ratio of 13C, making the signal for 13C much lower than that of a proton.
3.
Strong coupling to protons, although first order, gives complex multiplets
which often overlap, making peak assignments difficult.
These
limitations made the development of 13C NMR spectroscopy lag
sig-nificantly behind the development of 1H NMR. In the earliest
work the relatively weak sensitivity of 13C was a major stumbling
block and compounds specifically labeled with 13C had to be prepared
in order to obtain usable spectra. Today it is possible to obtain excellent 13C
spectra on natural abundance samples of <25
mg in less than 30 min. The hardware and software advances which have enabled
such progress to be made lie in three areas:
1. Improved signal detection
2. Fourier transform techniques
3.
Digital signal averaging
Suffice
it to say that modern NMR spectrometers are capable of obtaining 13C
spectra quickly and easily so that 13C NMR is now a routine tool for
structure identification.
These
instrumental improvements do not solve the problems of coupling between protons
and carbon that complicate 13C spectra, but other techniques have.
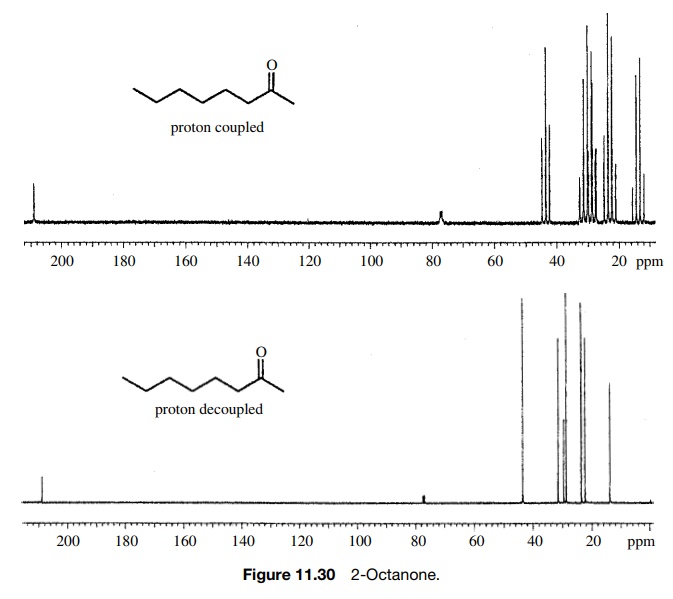
The proton-coupled 13C spectrum of 3-octanone demonstrates that pro-ton
– carbon coupling significantly complicates the spectrum due to the large
number of lines produced. From this spectrum, it is impossible to tell how many
carbons are present or what are their chemical shifts because of overlapping
mul-tiplets in the spectrum. To solve this problem, broad-band proton
decoupling is used to remove all proton – carbon couplings, and one is left
with proton decou-pled or fully decoupled spectra which have only singlet
absorptions for each carbon present. For example, 3-octanone has eight lines in
the fully decoupled 13C spectrum, as predicted by the fact that each
of the carbons is in a unique chemical environment and thus has a unique
chemical shift. A distinct advantage of 13C NMR is that 13C
absorbs over a range of ∼250 ppm (compared to
10 ppm for 1H). This means that each carbon can be distinguished by
a unique chem-ical shift. Thus it is possible to tell exactly how many
nonequivalent carbons are present in a molecule merely by counting the lines in
the fully decoupled spectrum (Figure 11.30). (The small three-line signal at
77.3δ is from the solvent CDCl3.
It appears in all 13C spectra run in CDCl3 and is
normally ignored.)
In
addition to the number of nonequivalent carbons present, the chemical shifts of
the carbons can reveal a great deal about the types of bonding patterns and
substituents which are present. Because of the great range of chemical shifts
observed for 13C (≈250
ppm), even small changes in the environment around carbon can result in a
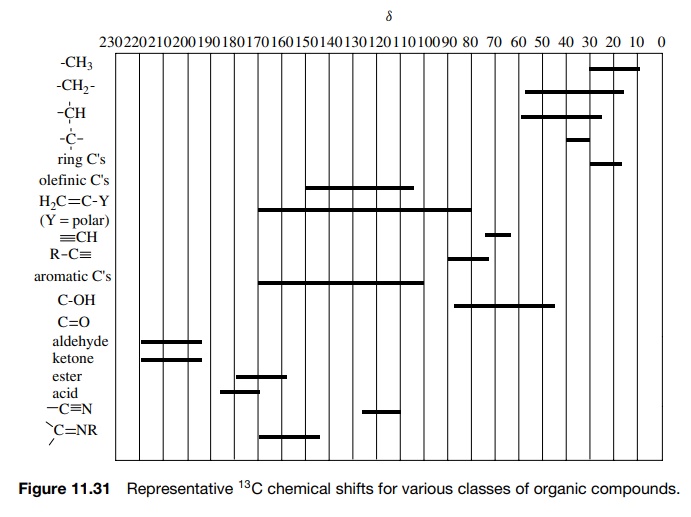
significant change in chemical shift. Figure 11.31 is a brief compilation of 13C
chemical shifts for representative classes of organic compounds. By assigning
the chemical shifts in many series of compounds, it has been possible to
develop correlation equations for calculating 13C chemical shifts
based on structural features present in the molecule. These correlation
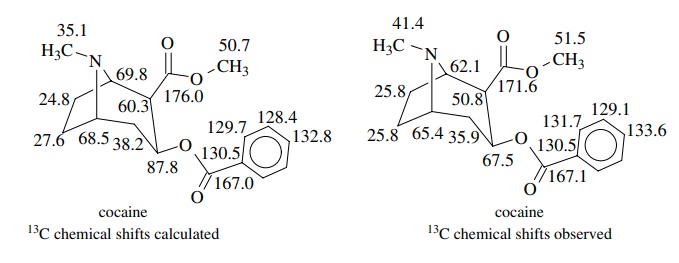
Thus it
is now routine to test possible structures by calculating 13C
chemical shifts and comparing them with the observed spectra. For example, the
calculated and observed 13C chemical shift values for cocaine are
seen to be in remarkable agreement for most of the carbons in this reasonably
complex compound.
The
value of fully decoupled 13C NMR spectra is primarily tied to
deter-mining how many nonequivalent carbons are present and their chemical
shifts. Unfortunately, the integrated areas of 13C signals are not
directly proportional to the numbers of carbons responsible for those signals
under most circumstances. Thus both n-heptane
and 4-(1-propyl) heptane have four signals in their 13C NMR spectra,
but it is not possible to determine if the ratio of different carbon types is 1
: 2 : 2 : 2 as expected for n-heptane
or the 1 : 3 : 3 : 3 expected for the branched compound.
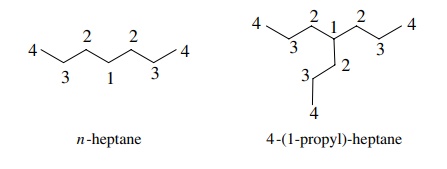
It
is possible to distinguish them based
on the chemical shift of C-4, which is calculated to be 36.5 ppm in n-heptane but 53.1 ppm in the branched
compound.
A
second difficulty of fully decoupled 13C NMR spectra is that the
connectiv-ity in the molecule is difficult to establish (except by chemical
shift correlation) because coupling patterns are absent. This dilemma is
partially resolved by the use of a technique called off-resonance decoupling.
In off-resonance decoupled 13C spectra, the carbons are coupled only
to those protons directly attached to them and the coupling is first order.
Thus quaternary carbons are singlets, methine carbons are doublets, methylene
carbons are triplets, and methyl carbons are quar-tets. It is possible to use
this information to establish proton – carbon connectivity, which can be used
to add protons to partial structures determined by 13C chemical
shift data.
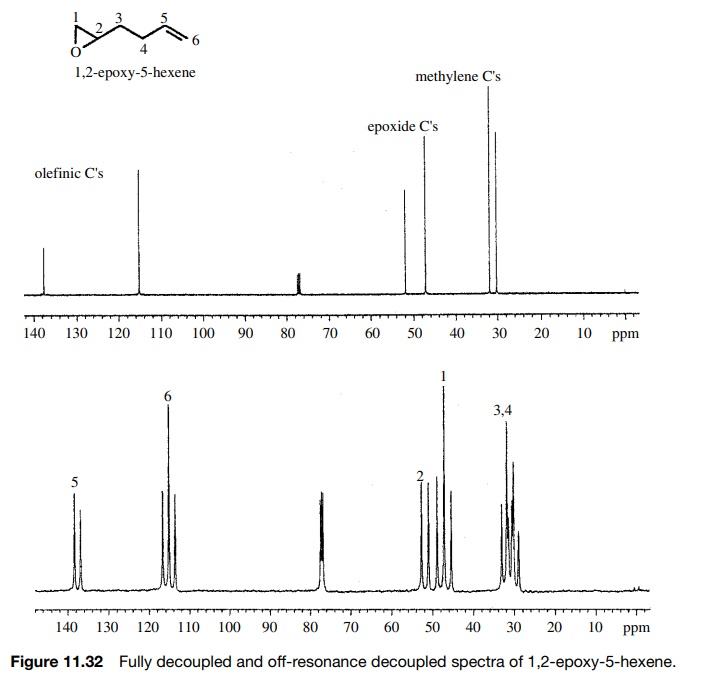
The
carbons of 1,2-epoxy-5-hexene can be assigned from the off-resonance decoupled
spectrum (Figure 11.32). In the fully decoupled spectrum it is clear that the
olefinic carbons (≈115 and 138δ) are distinct from the epoxide carbons
(≈47 and 52δ) and from the methylene carbons (≈30 and 32δ), but it is not possible to assign which is which. In the
off-resonance decoupled spectrum, both the olefinic and epoxide carbons are
distinguished by their splitting patterns from the numbers of directly attached
protons. The methylene carbons, however, are both triplets and cannot be
distinguished.
A
final feature of importance in 13C NMR spectra is the notion of
equiva-lency. Because some type of decoupling is normally done, either broad
band or off resonance, magnetic equivalency is not an issue in 13C
NMR, but chemi-cal equivalence remains an issue. If two carbon atoms share the
same chemical environment, then of course they will have the same chemical
shift. Thus it is important to recognize local or molecular symmetry elements.
In a previous
The internal plane of symme-try results in three equivalent pairs of
carbons in addition to the unique central carbon. Toluene (or any
monosubstituted benzene) has four signals for the aro-matic protons in addition
to the methyl carbon signal. The xylenes offer another example of equivalency.
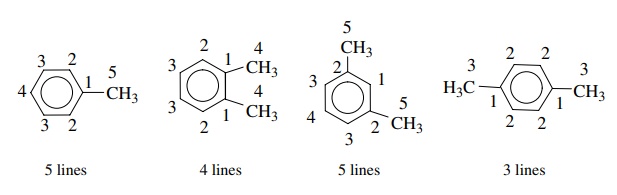
o-Xylene has four
signals, m-xylene has five signals,
and p-xylene has only 3. In general, the more symmetric is a
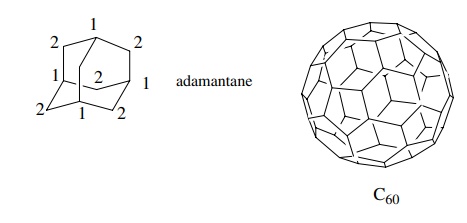
molecule, the fewer 13C signals it will have. For example adamantane
has only 2 absorptions and buckminsterfulluene (C60) has only a
single line in its 13C spectrum.
Thus
when the number of 13C signals is less than the number of carbon
atoms present in the molecule, there must be symmetry elements present that
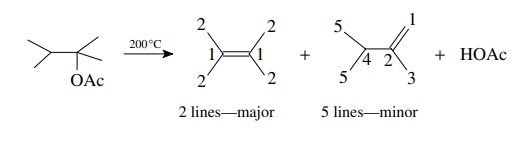
make some carbon atoms equivalent. The pyrolysis of 2-acetoxy-2.3-dimethylbutane
in a hot tube at 200◦
C gives two products which are both found to have the formula C6H12.
The major product has only two 13C absorptions while the minor
product has five 13C signals (Figure 11.33). Thus the major product
is likely to
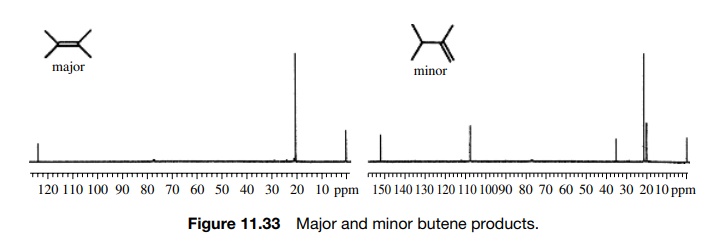
Note
that even the minor product has a pair of equivalent carbons giving rise to
five rather than six lines, but the symmetry is still significantly less than
that of the major olefin.
The
foregoing has been a brief introductory discussion of NMR which has
concentrated on some basic principles that are very useful in understanding the
technique. The actual practice of NMR today is much more advanced. The
incor-poration of Fourier transform techniques has revolutionized NMR
spectroscopy. All types of pulse sequences and two-dimensional (2D) techniques
have been developed to provide even greater structural detail than has been
discussed above. A discussion of such techniques belongs in a more specialized
text, but it must be remembered that while these techniques are faster, more
sensitive, and much more sophisticated, they are still largely based on the
principles presented here, as is the interpretation of the results.
Related Topics
