Validation and In-Process Monitoring of Sterilization Procedures
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Sterilization Procedures And Sterility Assurance
There are several definitions of ‘validation’ but, in simple terms, the word means demonstrating that a process will consistently produce the results that it is intended to. Thus, with respect to sterile products, validation would be necessary for each of the individual aspects of the manufacturing process.
VALIDATION
AND IN-PROCESS MONITORING OF
STERILIZATION PROCEDURES
There are several definitions of ‘validation’ but, in simple terms, the word means demonstrating that a process
will consistently produce
the results that it is intended to. Thus, with
respect to sterile
products, validation would be necessary for
each of the
individual aspects of the
manufacturing process,
e.g. environmental monitoring, raw materials quality
assessment, the sterilization process itself and the sterility testing
procedure. Of these,
it is the sterilization process that is likely
to be subject to the most
detailed and complex validation procedures, and these will be used to exemplify the factors to be considered. A typical validation procedure for a steam sterilization
process is likely to incorporate most, or all, of the following features:
•
Calibration and testing of all the physical instruments used to monitor the process, e.g.
thermocouples, pressure gauges and timers
•
Production of evidence that
the steam is of the desired
quality (e.g. that
the chamber temperature is that expected for pure steam at the measured pressure)
•
Conduct of leak tests and steam penetration tests using both an empty chamber
and a chamber filled with the product to be sterilized in the intended load conformation
•
Use of
biological indicators either alone or in combination with bioburden organisms to demonstrate that the sterilization cycle is capable of
producing an acceptable level of
sterility assurance under ‘worst case’ conditions
•
Production of data to demonstrate repeatability of the above (typically for three runs)
•
Testing of
software associated with parametric and operational monitoring
• Comprehensive documentation of all of these aspects.
There are different approaches to the demonstration of adequate sterility
assurance in steam sterilization depending upon the thermostability and knowledge of the pre-sterilization bioburden. Where
the product is known to be stable, an overkill approach may be adopted in which biological indicators containing 106 test organisms are inactivated in half the proposed
exposure time (thus achieving a 12-log reduction and a sterility assurance level of 10−6 in the full
exposure period).
For a marginally thermostable product the cycle could be validated on the basis
of measurements of the
worst case bioburden level and
the heat resistance of the known bioburden organisms; such an approach would necessitate rigorous control of
the bioburden during routine manufacturing. In the UK, biological indicators are used primarily
in validation rather than routine
monitoring of heat sterilization processes, although their use in routine manufacturing may be required
in other countries. Chemical indicators of sterilization are more convenient to use than biological indicators, but as they provide no direct
measure of the efficacy of the
process in terms of microbial killing they are
considered to be less
useful. In certain
instances these are
no longer routinely used. Physical measurements of
temperature, pressure, time, relative
humidity, etc. are of such fundamental importance to the assurance of sterility that records of these parameters are retained for each batch of
sterilized product.
a)
Physical Indicators
In heat sterilization processes, a temperature record
is made of each
sterilization cycle with
both dry and
moist heat (i.e.
autoclave) sterilizers; this chart/digital record forms part of the batch documentation and is compared against a master temperature record (MTR). It is recommended that the temperature be taken at the coolest
part of the loaded sterilizer. Further information on heat distribution and penetration within a sterilizer can be gained by the use of thermocouples placed
at selected sites in the chamber or inserted directly
into test packs
or bottles. For gaseous sterilization procedures, elevated temperatures are monitored for each sterilization cycle by temperature probes,
and routine leak tests are performed
to ensure gas-tight seals. Pressure and humidity measurements are recorded. Gas concentration is measured independently of pressure rise, often by reference to weight of gas used. In radiation sterilization, a plastic (often Perspex) dosimeter which gradually darkens
in proportion to the radiation
absorbed gives an accurate measure of the radiation dose and
is considered to be the
best technique currently available for following
the radio sterilization process.
Sterilizing filters
are subject to a bubble
point pressure test, which is a technique employed for determining the pore size of filters, and may also
be used to check the integrity of certain
types of filter device (membrane and sintered glass)
immediately after use.
The principle of the
test is that
the wetted filter,
in its assembled unit, is subjected to an increasing air or nitrogen gas pressure differential. The pressure difference recorded when the first bubble of gas breaks away from the filter is related to the maximum
pore size. When
the gas pressure is further increased slowly,
there is a general eruption of bubbles over
the entire surface. The pressure difference here
is related to the mean
pore size. A pressure
differential below the expected value would
signify a damaged or
faulty filter. A modification to this test for membrane filters
involves measuring the diffusion of gas through
a wetted filter
at pressures below the bubble point pressure
(diffusion rate test);
a faster diffusion rate than expected would again indicate
a loss of filter integrity. In addition, a filter is considered
ineffective when
an unusually rapid
rate of filtration occurs.
Efficiency testing
of HEPA filters used for the supply
of sterile air to aseptic
workplaces is normally achieved
by the generation upstream of dioctylphthalate (DOP)
or sodium chloride
particles of known dimension followed by detection in downstream filtered air. Retention efficiency is recorded as the percentage of particles removed under defined test conditions. Microbiological tests are not normally done.
b) Chemical Indicators
Chemical monitoring of a sterilization process is based on
the ability of heat, steam,
sterilant gases and ionizing
radiation to alter the chemical and/or
physical characteristics of a variety of chemical substances. Ideally, this change should take place
only when satisfactory conditions for sterilization prevail,
thus confirming that the
sterilization cycle has been successfully completed. In practice,
however, the ideal indicator
response is not always achieved
and so a necessary distinction is made between (1) those chemical
indicators which integrate several sterilization parameters (i.e.
temperature, time and saturated steam) and closely
approach the ideal;
and those which measure
only one parameter
and consequently can only be used to distinguish processed from unprocessed articles. Thus, indicators which
rely on the melting of a chemical substance show that the temperature has been attained
but not necessarily maintained.
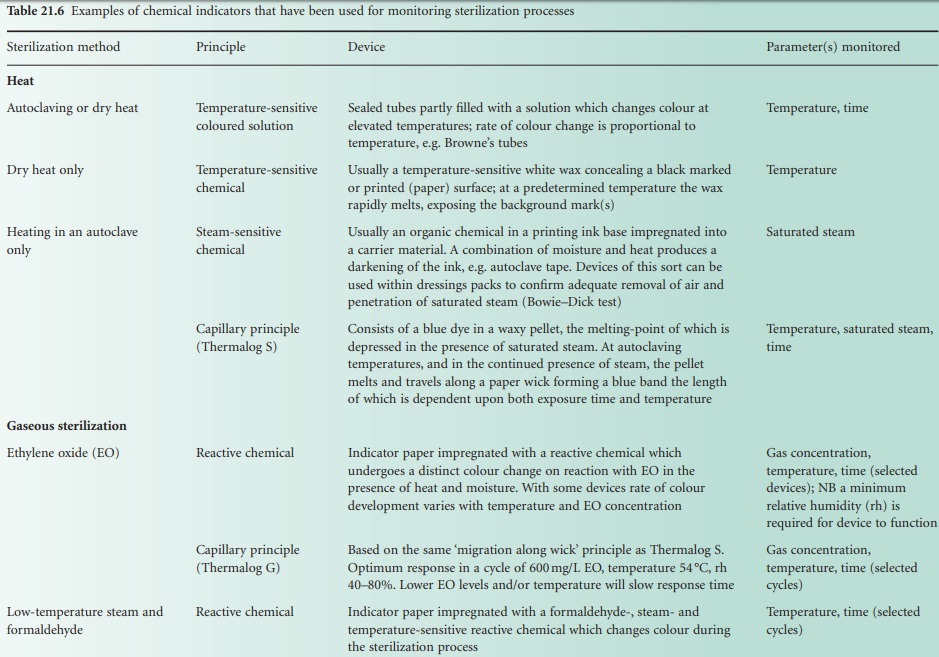

Chemical indicators
generally undergo melting or colour changes, the relationship of this change
to the sterilization
process being influenced by the design of the test
device (Table 21.6). It must be remembered, however,
that the changes
recorded do not necessarily correspond to microbiological sterility
and consequently the devices should never be employed as sole indicators in a sterilization process. Nevertheless, when included in strategically
placed containers or packages, chemical
indicators are valuable monitors of the conditions prevailing at the coolest or most inaccessible parts of a sterilizer.
c) Biological Indicators
Biological indicators (BIs) for use
in thermal, chemical or radiation
sterilization processes consist of standardized bacterial spore
preparations which are usually in the form either of suspensions in water or culture medium
or of spores dried
on paper, aluminium or plastic carriers. As with chemical
indicators, they are usually placed
in dummy packs
located at strategic sites in the
sterilizer. Alternatively,
for gaseous sterilization these may also be placed within a tubular helix (Line–Pickerill) device. After the sterilization process, the aqueous
suspensions or spores
on carriers are aseptically transferred to
an appropriate nutrient medium,
which is then
incubated and periodically examined for signs of growth. Spores
of stearothermophilus in sealed ampoules of culture medium are used for steam sterilization monitoring, and these may
be incubated directly at 55 °C;
this eliminates the need for an aseptic
transfer. Aseptic transfers are also avoided by the use of self-contained units where the spore
strip and nutrient medium are present in the same device
ready for mixing after use.
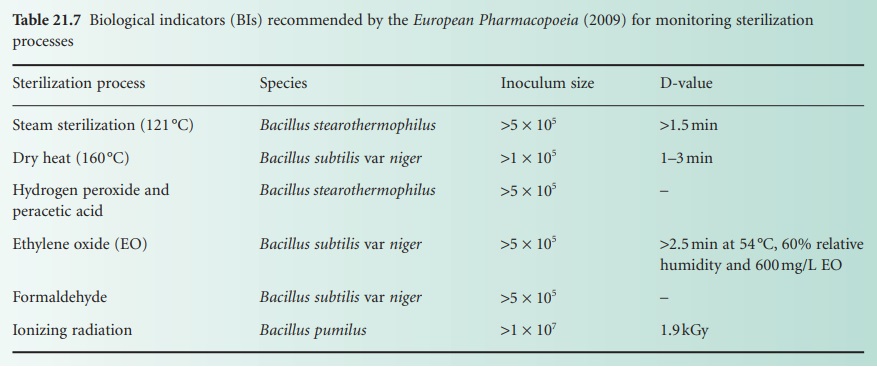
The bacterial species to be used in a BI must be selected
carefully, as it must be non-pathogenic and should possess above-average resistance to the particular sterilization process. Resistance is adjudged from the spore destruction curve obtained upon
exposure to the
sterilization process; recommended BI spores
and their decimal reduction times (D-values) are shown in Table 21.7. Great
care must be taken in the preparation and storage of BIs
to ensure a standardized response to sterilization processes. Indeed,
while certainly offering the most direct method
of monitoring sterilization processes, it should
be realized that BIs may be less reliable monitors than physical
methods and they are not recommended for routine
use, except in the case of gaseous
sterilization.
One of the long-standing criticisms of BIs is that the
incubation period
required in order
to confirm a satisfactory sterilization process imposes an undesirable delay
on the release
of the product. This problem
has been overcome, with respect to steam sterilization at least, by the use of a detection
system in which a spore enzyme, α-glucosidase (reflective of spore viability), converts a non-fluorescent substrate into a fluorescent product
in as little as 1 hour.
Filtration sterilization requires a different
approach from biological monitoring, the test effectively measuring the ability of a filter
to produce a sterile filtrate from a culture of a suitable organism. For this purpose,
Serratia marcescens, a small Gram-negative rod-shaped bacterium (minimum dimension 0.5 μm), has been
used for filters of 0.45 μm pore size,
and a more rigorous test
involving Brevundimonas
diminuta (formerly Pseudomonas diminuta) having a minimum dimension of 0.3 μm is
applied to filters of 0.22 μm pore size.
The latter filters
are defined as those capable of completely removing
Brev. diminuta from suspension. In this test, using this organism, a realistic
inoculum level must be adopted,
as the probability of
bacteria appearing in the filtrate
rises as the number of Brev. diminuta
cells in the test challenge increases; a standardized inoculum size of 107 cells
cm−2 is normally
employed. The extent of the passage of this
organism through membrane filters
is enhanced by increasing the filtration pressure. Thus, successful sterile filtration depends markedly
on the challenge conditions.
Such
tests are used as part of the filter manufacturer’s characterization and quality assurance process, and a user’s initial validation procedure. They are not employed as a test of filter
performance in use.
Related Topics
