Angiotensin
| Home | | Pharmacology |Chapter: Essential pharmacology : Drugs Affecting Renin-Angiotensin System And Plasma Kinins
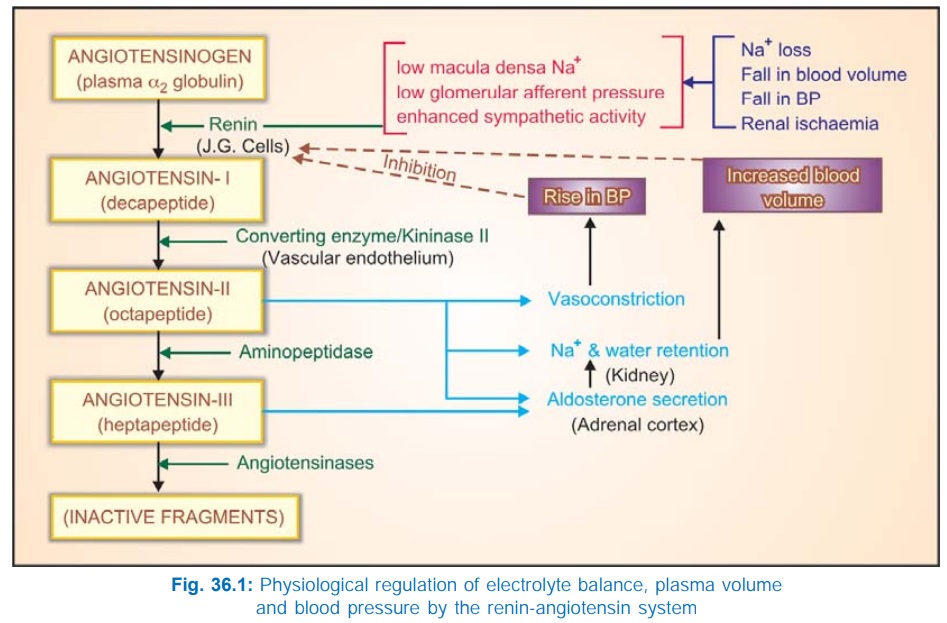
Angiotensin-II (AII) is an octapeptide generated in the plasma from a precursor plasma α2 globulin, and is involved in electrolyte, blood volume and pressure homeostasis. Pressor action of kidney extracts was known since the turn of the 19th century.
ANGIOTENSIN
Angiotensin-II (AII) is an octapeptide
generated in the plasma from a precursor
plasma α2 globulin, and is
involved in electrolyte, blood volume and pressure homeostasis. Pressor action
of kidney extracts was known since the turn of the 19th century. The active
material was termed ‘Renin’. In the 1940s renin was shown to be an enzyme which
acted indirectly by producing a pressor principle from plasma protein.
Subsequently, it became clear that the product of renin action was an inactive
decapeptide angiotensinI (AI) which
was converted to the active octapeptide AII by an angiotensin converting enzyme
(ACE). The renin-angiotensin system (RAS) has attracted considerable attention
in the recent years, particularly after the development of ACE inhibitor captopril.
Circulating Renin-Angiotensin System
The generation and metabolism of AII in circulation
is depicted in Fig. 36.1. Normally, the amount of renin in plasma acts as the
limiting factor for AII generation. The plasma t½ of renin is 15 min. The
biological potency of AI is only 1/100 that of AII, but it is rapidly converted
into the latter by ACE which is a dipeptidyl
carboxypeptidase located primarily on the luminal surface of vascular
endothelial cells (especially in lungs). Circulating AII also has a very short
t½ (1 min); the first degradation product termed AngiotensinIII (AIII) is 2–10 times less potent than AII, except in
stimulating aldosterone secretion, in which it is equipotent. AIII is further
acted upon by a variety of peptidases, collectively termed angiotensinases, to
inactive fragments.
Tissue (Local) Renin-Angiotensin Systems
Apart from the AII generated in circulation as
described above, blood vessels capture circulating renin and angiotensinogen
and produce AII within or at the surface of their wall (extrinsic local RAS). Many tissues, especially heart, blood vessels,
brain, kidneys, adrenals possess all components of the renin-angiotensin system
and generate AII inside their cells (intrinsic
local RAS). Thus, local renin-angiotensin systems appear to operate in
several organs in addition to the circulating one.
Actions
CVS
The most prominent
action of AII is vasoconstriction—produced
directly as well as by enhancing Adr/NA release from adrenal medulla/adrenergic
nerve endings and by increasing central sympathetic outflow. Vasoconstriction
involves arterioles and venules and occurs in all vascular beds. However, it is
less marked in cerebral, skeletal muscle, pulmonary and coronary vessels. AII induced vasoconstriction
promotes movement of fluid from vascular to extravascular compartment. BP rises
acutely. As a pressor agent, AII is much more potent than NA. No tachyphylaxis
is seen in the pressor action of AII; rather long-term infusion of low
concentration of AII produces sustained rise in BP by its renal effects promoting
salt and water reabsorption, as well as by enhancing endothelin generation.
AII increases force of
myocardial contraction by promoting Ca2+ influx. Though, it can
increase heart rate by enhancing sympathetic activity, reflex bradycardia
predominates in the intact animal. Cardiac output is often reduced and cardiac
work is increased (due to rise in peripheral resistance). In contrast to NA, AII
does not activate latent pacemakers—little arrhythmogenic propensity.
AII acting on a chronic basis induces hypertrophy, hyperplasia
and increased intercellular matrix production in the myocardium and vascular
smooth muscle by direct cellular effects involving expression of protooncogenes
and transcription of several growth factors. Indirectly, volume overload and
increased t.p.r. caused by AII contributes to the hypertrophy and remodeling
(abnormal redistribution of muscle mass) in heart and blood vessels. Long
standing hypertension increases vessel wall + intimal thickness and causes
ventricular hypertrophy. Fibrosis and dilatation of infarcted area with
hypertrophy of the non-infarcted ventricular wall is seen after myocardial infarction.
Progressive cardiac myocyte death and fibrotic transformation occurs in CHF.
These changes are important risk factors for cardiovascular morbidity and mortality.
ACE inhibitor therapy retards/reverses many of these changes imparting a
pivotal role to AII in vascular and ventricular hypertrophy, apoptosis and
remodeling.
Smooth Muscles
AII contracts many visceral
smooth muscles in vitro, but in vivo effects are insignificant.
Adrenal Cortex
AII and AIII are
trophic to the zona glomerulosa of
the adrenal cortex— enhance synthesis and release of aldosterone which acts on
distal tubule to promote Na+ reabsorption and K+/H+
excretion. These effects are exerted at concentrations lower than those
required to cause vasoconstriction.
Kidney
In addition to
exerting indirect effect on kidney through
aldosterone, AII promotes Na+/H+ exchange in proximal
tubule → increased Na+,
Cl– and HCO3¯ reabsorption. Further, it reduces renal blood flow and
produces intrarenal haemodynamic effects which normally result in Na+
and water retention. However, an opposite effect has been observed in
cirrhotics and renovascular disease patients.
CNS
It has been noted that
systemically administered AII can
gain access to certain periventricular areas of the brain to induce drinking
behaviour and ADH release—both of which would be conducive to plasma volume
expansion. It also increases central sympathetic outflow —contributes to the
pressor response.
Peripheral Sympathetic Structures
AII enhances sympathetic activity by peripheral
action as well. It releases Adr from adrenal medulla, stimulates autonomic
ganglia and increases the output of NA from adrenergic nerve endings.
Angiotensin Receptors And Transducer Mechanisms
Specific angiotensin
receptors are present on the surface
of target cells. Two subtypes (AT1 and AT2) have been differentiated
pharmacologically: Losartan is a
selective AT1 antagonist, while PD 123177 is a selective AT2 antagonist. Both
subtypes are G-protein coupled receptors. However, all known effects of AII
appear to be mediated by AT1 receptor.
The AT2
receptor is abundantly expressed in foetal tissues. In adults, it has been
demonstrated in vascular endothelium, adrenal medulla, kidney and some brain
areas. The functional role of AT2 receptor is not clearly defined,
but is generally opposite to that of AT1 receptor. Activation of AT2
receptor causes NO-dependent vasodilatation, promotes apoptosis, myocardial
fibrosis and inhibits cell proliferation.
The AT1 receptor
utilizes different transducer mechanisms in different tissues. The phospholipase
C–IP3/DAG–intracellular Ca2+ release mechanism underlies vascular
and visceral smooth muscle contraction by activating myosin light chain kinase
(MLCK). In addition, membrane Ca2+ channels are activated. Enhanced
Ca2+ movement also induces aldosterone synthesis/release, cardiac
inotropy, depolarization of adrenal medullary/autonomic ganglionic cell
resulting in CA release/ sympathetic discharge. DAG activates protein kinase C
(PKC) which phosphorylates several intracellular proteins and augments the
above responses as well as participates in promotion of cell growth. In liver
and kidney, AII inhibits adenylyl cyclase. The intrarenal homeostatic action
involves phospholipase A2 activation and PG/LT production.
In many tissues,
especially myocardium, vascular smooth muscle and fibroblasts, AT1 receptor
also mediates long-term effects of AII on cell growth. AII activates MAP
kinase, TAK2 tyrosine protein kinase and PKC which together enhance expression
of protooncogenes, transcription factors and growth factors. As a result, cell
growth is promoted and more intercellular matrix is synthesized.
Pathophysiological Roles
Mineralocorticoid
Secretion
There is no doubt that AII (also AIII) is the physiological stimulus
for aldosterone secretion from adrenal cortex. It also exerts trophic influence
on the glomerulosa cells so that effects are augmented under conditions which
persistently raise AII levels.
Electrolyte,
Blood Volume And Pressure Homeostasis
The RAS plays an
important role in maintaining
electrolyte composition and volume of extracellular fluid (see Fig. 36.1). Changes that lower blood volume or pressure, or
decrease Na+ content induce renin release by—
1.
Decreasing tension in the afferent glomerular
arterioles: the intrarenal baroreceptor
pathway: possibly operates through increasing local production of prostaglandins (PGs).
2.
Low Na+ concentration in the
tubular fluid sensed by macula densa cells: the
macula densa pathway. It has been
found that COX2 and neuronal nitric
oxide synthase (n-NOS) are induced in macula densa cells by Na+
depletion → release of PGE2 and
PGI2 is enhanced both due to increased amount of COX2 as well as its activation
by NO. The locally released PGs act on juxtaglomerular cells to promote renin
secretion.
3.
Baroreceptor and other reflexes which increase
sympathetic impulses to JG cells— activated through β1 receptors: the β adrenoceptor pathway.
Increased renin is
translated into increased plasma AII which produces acute rise in BP by vasoconstriction,
and more long-lasting effects by directly as well as indirectly increasing Na+
and water reabsorption in the kidney. Rise in BP in turn inhibits renin release
: the long-loop negative feedback mechanism. It has been recently
shown that AII can be formed within
the kidney and exerts important local regulatory effects. A short-loop negative feedback mechanism operates
within the kidney : activation of AT1
receptors on JG cells inhibits renin release. Long-term stabilization of BP despite
varying salt and water intake appears to be achieved through these mechanisms.
The mechanisms of regulation of renin release have important
pharmacological implications:
·
ACE inhibitors and AT1 antagonists enhance
renin release by interfering with both the shortloop and longloop negative
feedback mechanisms.
· Vasodilators and diuretics stimulate renin
release by lowering BP.
· Loop diuretics increase renin production by
reducing entry of Na+ into macula densa cells.
· Central sympatholytics and β blockers decrease
renin release by depressing the β adrenoceptor pathway.
· NSAIDs, including selective COX2 inhibitors,
and nNOS inhibitors decrease renin release by inhibiting PG production → cause Na+
and water retention.
Development
of Hypertension
The RAS is directly involved in renovascular
hypertension: plasma renin activity (PRA) is raised in most patients. In
essential hypertension also it appears to have a permissive role, though PRA
may be raised or low. Since ACE inhibitors consistently lower BP in
hypertensives, the involvement of this system appears to be more widespread. A
positive correlation between circulating angiotensinogen levels and essential
hypertension has also been found. Several genetic evidences point to causation
of pregnancy-induced hypertension (preeclampsia) by production of AT1
receptor agonistic autoantibodies. The role of AII in hypertrophy/remodeling of
heart and blood vessels is now well recognized (see above).
Secondary Hyperaldosteronism
The RAS is instrumental in the
development of secondary hyperaldosteronism.
CNS
AII can be formed
locally in the brain and may function as
transmitter or modulator. Regulation of thirst, hormone release and sympathetic
flow may be the responses mediated.
AII is not available commercially,
and not used clinically.
Inhibition Of Renin-Angiotensin System
It can be achieved by:
1.
Sympathetic blockers (β blockers, adrenergic
neurone blockers, central sympatholytics)— decrease renin release.
2.
Renin inhibitory peptides and renin specific
antibodies block renin action—interfere with generation of AI from
angiotensinogen (rate limiting step).
3.
Angiotensin converting enzyme inhibitors—
prevent generation of the active principle AII.
4.
Angiotensin receptor (AT1) antagonists— block
the action of AII on target cells.
5.
Aldosterone antagonists—block mineralocorticoid
receptors.
Related Topics
