Antiepileptic Drugs
| Home | | Pharmacology |Chapter: Essential pharmacology : Antiepileptic Drugs
Phenobarbitone was the first efficacious antiepileptic introduced in 1912. The same may apply to anticonvulsant action. GABAA receptor mediated synaptic inhibition appears to be most important.
ANTIEPILEPTIC DRUGS
CLASSIFICATION
1. Barbiturate
Phenobarbitone
2. Deoxybarbiturate
Primidone
3. Hydantoin
Phenytoin
Fosphenytoin
4. Iminostilbene
Carbamazepine
Oxcarbazepine
5. Succinimide
Ethosuximide
6. Aliphatic carboxylic acid
Valproic acid
(sodium valproate)
Divalproex
7. Benzodiazepines
Clonazepam
Diazepam
Lorazepam
Clobazam
8. Phenyltriazine
Lamotrigine
9) Cyclic GABA analogue
Gabapentin
10. Newer drugs
Vigabatrin
Topiramate
Tiagabine
Zonisamide
Levetiracetam
The oxazolidinedione derivative Trimethadione, acetylurea Phenacemide and carbonic anhydrase inhibitor Acetazolamide are anticonvulsants no longer used due to their toxicity or relative inefficacy.
Chemistry
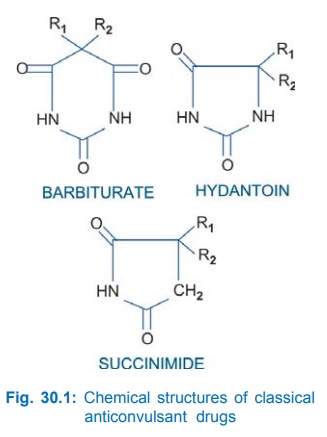
Most of the older anticonvulsants have close structural similarity. This is depicted in Fig. 30.1. However, benzodiazepines, carbamazepine, valproic acid and the newer drugs are chemically diverse. Presence of a phenyl substitution confers activity against tonicclonic seizures.
Phenobarbitone
Phenobarbitone was the first efficacious antiepileptic introduced in 1912. The same may apply to anticonvulsant action. GABAA receptor mediated synaptic inhibition appears to be most important. However, phenobarbitone has specific anticonvulsant activity which is not entirely dependent on general CNS depression. Quantitative differences in the different facets of action (GABA-facilitatory, GABA-mimetic, anti-glutamate, Ca2+ entry reduction) have been noted for phenobarbitone compared to hypnotic barbiturates. The higher anticonvulsant: hypnotic ratio of phenobarbitone may be due to its minimal effect on Ca2+ channels and glutamate release compared to hypnotic barbiturates. With continued use of phenobarbitone sedation wanes off but not anticonvulsant action. It has a wide spectrum of anticonvulsant property—raises seizure threshold as well as limits spread and suppresses kindled seizures.
Phenobarbitone has slow oral absorption and a long plasma t½ (80–120 hours), is metabolized in liver as well as excreted unchanged by kidney. Steadystate concentrations are reached after 2–3 weeks, and a single daily dose can be used for maintenance.
The major drawback of phenobarbitone as an antiepileptic is its sedative action. Long term administration (as needed in epilepsy) may produce additional side effects like—behavioral abnormalities, diminution of intelligence, impairment of learning and memory, hyperactivity in children, mental confusion in older people.
Rashes, megaloblastic anaemia and osteomalacia (similar to that with phenytoin) occur in some patients on prolonged use.
Uses
Phenobarbitone is one of the cheapest and least toxic antiepileptics.
It has broad spectrum efficacy in generalized tonicclonic (GTC), simple partial (SP) and complex partial (CP) seizures: 60 mg 1–3 times a day in adults; in children (3–6 mg/kg/day); However, it has become less popular than carbamazepine, phenytoin or valproate.
Status epilepticus: Phenobarbitone may be injected i.m. or i.v. but response is slow to develop. It is not effective in absence seizures.
GARDENAL 30, 60 mg tabs, 20 mg/5 ml syr; LUMINAL 30 mg tab, PHENOBARBITONE SODIUM 200 mg/ml inj.
Primidone
A deoxybarbiturate, converted by liver to phenobarbitone and phenylethyl malonamide (PEMA). Activity is mainly due to these active metabolites because t½ of primidone (6–14 hr) is less than that of its active metabolites. About 1/3 primidone is excreted unchanged by kidney. Dose to dose primidone is less potent, but antiepileptic efficacy is similar to phenobarbitone. It is infrequently used now in GTCS and partial epilepsy, mainly as an adjuvant to phenytoin or carbamazepine.
Some cases of myoclonic epilepsy respond. Adverse effects are similar to phenobarbitone. In addition, anaemia, leukopenia, psychotic reaction and lymph node enlargement occur rarely.
Dose: 250–500 mg BD, children 10–20 mg/kg/day.
MYSOLINE 250 mg tab.
Phenytoin (Diphenylhydantoin)
It was synthesized in 1908 as a barbiturate analogue, but shelved due to poor sedative property. Its anticonvulsant activity was specifically tested in 1938 and since then it is a major antiepileptic drug.
Phenytoin is not a CNS depressant; some sedation occurs at therapeutic doses, but this does not increase further with dose; rather toxic doses produce excitement and muscular rigidity. The most outstanding action is abolition of tonic phase of maximal electroshock seizures, with no effect on or prolongation of clonic phase. It limits spread of seizure activity. Threshold for PTZ convulsions is not raised. Tonicclonic epilepsy is suppressed but paroxysmal focal EEG discharge and ‘aura’ persist.
Mechanism Of Action
Phenytoin has a stabilizing influence on neuronal membrane—prevents repetitive detonation of normal brain cells during ‘depolarization shift’ that occurs in epileptic patients and consists of a synchronous and unusually large depolarization over which action potentials are superimposed. This is achieved by prolonging the inactivated state of voltage sensitive neuronal Na+ channel (Fig. 30.2) that governs the refractory period of the neurone. As a result high frequency discharges are inhibited with little effect on normal low frequency discharges which allow Na+ channels to recover even when inactivation is prolonged. This effect has been noted at therapeutic concentration of phenytoin, while other effects like reduction in Ca2+ influx, inhibition of glutamate and facilitation of GABA responses have been demonstrated at higher/toxic concentrations. Intracellular accumulation of Na+ that occurs during repetitive firing is prevented.
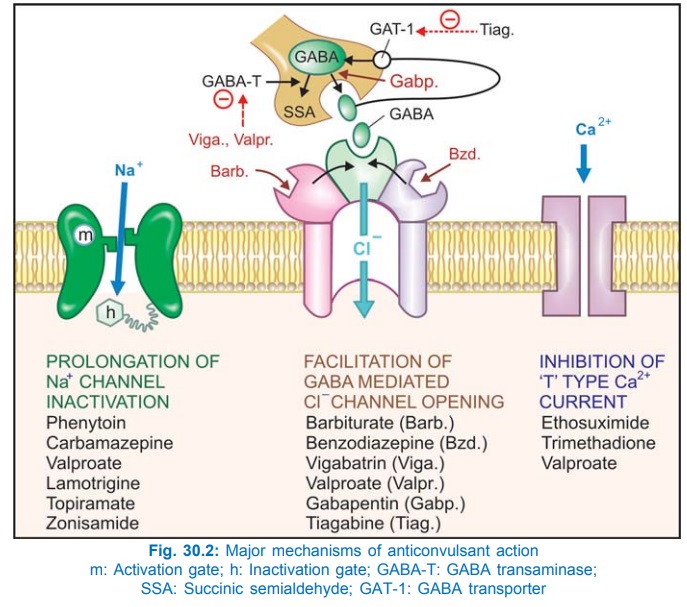
Therapeutic concentrations have no effect on resting membrane potential: normal synaptic transmission is not impaired. Phenytoin, in contrast to phenobarbitone and valproate, does not interfere with kindling. Its ability to selectively inhibit high frequency firing confers efficacy in trigeminal neuralgia and cardiac arrhythmias as well.
Pharmacokinetics
Absorption of phenytoin by oral route is slow, mainly because of its poor aqueous solubility. Bioavailability of different market preparations may differ. It is widely distributed in the body and is 80–90% bound to plasma proteins.
Phenytoin is metabolized in liver by hydroxylation and glucuronide conjugation. The kinetics of metabolism is capacity limited; changes from first order to zero order over the therapeutic range—small increments in dose produce disproportionately high plasma concentrations. The t½ (12–24 hours) progressively increases (upto 60 hr) when plasma concentration rises above 10 μg/ml as metabolizing enzymes get saturated. Monitoring of plasma concentration is very helpful in tailoring dosage. Only 5% unchanged phenytoin is excreted in urine.
Adverse Effects
These are numerous; some occur at therapeutic plasma concentration after prolonged use, while others are a manifestation of toxicity due to overdose.
At Therapeutic Levels
• Gum hypertrophy: Commonest (20% incidence), more in younger patients and is due to overgrowth of gingival collagen fibres. This can be minimized by maintaining oral hygiene.
• Hirsutism, coarsening of facial features (troublesome in young girls), acne.
• Hypersensitivity reactions are—rashes, DLE, lymphadenopathy; neutropenia is rare but requires discontinuation of therapy.
• Megaloblastic anaemia: phenytoin decreases folate absorption and increases its excretion.
• Osteomalacia: phenytoin desensitizes target tissues to vit D and interferes with calcium metabolism.
• It can inhibit insulin release and cause hyperglycaemia.
• Used during pregnancy—can produce foetal hydantoin syndrome (hypoplastic phalanges, cleft palate, hare lip, microcephaly), which is probably caused by its areneoxide metabolite.
At High Plasma Levels (Dose Related Toxicity)
• Cerebellar and vestibular manifestations: ataxia, vertigo, diplopia, nystagmus are the most characteristic features.
• Drowsiness, behavioral alterations, mental confusion, hallucinations, disorientation and rigidity.
• Epigastric pain, nausea and vomiting: minimised by taking the drug with meals.
• Intravenous injection can cause local vascular injury → intimal damage and thrombosis of the vein → edema and discolouration of the injected limb. Rate of injection should not exceed 50 mg/min.
• Fall in BP and cardiac arrhythmias occur only on i.v. injection.
Interactions
Phenobarbitone competitively inhibits phenytoin metabolism, while by enzyme induction both enhance each other’s degradation—unpredictable overall interaction.
• Carbamazepine and phenytoin increase each other’s metabolism.
• Valproate displaces protein bound phenytoin and decreases its metabolism: plasma level of unbound phenytoin increases.
• Chloramphenicol, isoniazid, cimetidine, dicumarol and warfarin inhibit phenytoin metabolism—can precipitate its toxicity.
• Phenytoin competitively inhibits warfarin metabolism.
• Phenytoin induces microsomal enzymes and increases degradation of steroids (failure of oral contraceptives), digitoxin, doxycycline, theophylline.
• A number of acidic drugs displace it from protein binding sites but this also enhances phenytoin clearance—concentration of free form does not change much.
• Sucralfate binds phenytoin in g.i. tract and decreases its absorption.
Uses
Phenytoin is a first line antiepileptic drug for—
• Generalized tonic-clonic, simple and complex partial seizures. It is ineffective in absence seizures.
Dose: 100 mg BD, maximum 400 mg/day; Children 5–8 mg/kg/day.
• Status epilepticus: occasionally used by slow i.v. injection (see later).
• Trigeminal neuralgia: second choice drug to carbamazepine.
DILANTIN 25 mg, 100 mg cap., 100 mg/4 ml oral suspension, 100 mg/2 ml inj; EPSOLIN 100 mg tab, 100 mg/2 ml inj; EPTON 50, 100 mg tab, 25 mg/ml syr; FENTOINER 100 mg extended release cap.
Fosphenytoin
This water soluble prodrug of phenytoin has been introduced to overcome the difficulties in i.v. administration of phenytoin in status epilepticus. In the body, it is rapidly converted to phenytoin; its doses are expressed as phenytoin equivalents (PE). On i.v. injection it is less damaging to the intima; few vascular complications are produced; it can be injected at a faster rate (150 mg/min). While phenytoin cannot be injected in a drip of glucose solution, fosphenytoin can be injected with both saline and glucose.
FOSOLIN 50 mg/ml in 2 ml, 10 ml inj.
Carbamazepine
Chemically related to imipramine, it was introduced in the 1960s for trigeminal neuralgia; is now a first line antiepileptic drug. Its pharmacological actions resemble phenytoin, but important differences have been noted in experimental studies. Carbamazepine modifies maximal electroshock seizures as well as raises threshold to PTZ and electroshock convulsions. It also inhibits kindling. Though its action on Na+ channels (prolongation of inactivated state) is similar to phenytoin, the profile of action on neuronal systems in brain is different.
Carbamazepine exerts a lithiumlike therapeutic effect in mania and bipolar mood disorder. It also has antidiuretic action, probably by enhancing ADH action on renal tubules.
Pharmacokinetics
Oral absorption of carbamazepine is slow and variable because of poor water solubility. It is 75% bound to plasma proteins and metabolized in liver by oxidation to an active metabolite (1011 epoxy carbamazepine) as well as by hydroxylation and conjugation to inactive ones. It is a substrate as well as inducer of CYP3A4 and other drug metabolizing enzymes. Initially its plasma t½ is 20–40 hours but, decreases to 10–20 hr on chronic medication due to autoinduction of metabolism.
Adverse Effects
Carbamazepine produces dose-related neurotoxicity—sedation, dizziness, vertigo, diplopia and ataxia. Vomiting, diarrhoea, worsening of seizures are also seen with higher doses. Acute intoxication causes coma, convulsions and cardiovascular collapse.
Hypersensitivity reactions are rashes, photosensitivity, hepatitis, lupus like syndrome, rarely agranulocytosis and aplastic anaemia. Some degree of leucopenia due to hypersensitivity is more common.
Water retention and hyponatremia can occur in the elderly because it enhances ADH action. Increased incidence of minor foetal malformations has been reported. Its combination with valproate doubles teratogenic frequency.
Interactions
Carbamazepine is an enzyme inducer; can reduce efficacy of haloperidol, oral contraceptives, lamotrigine and topiramate. Metabolism of carbamazepine is induced by phenobarbitone, phenytoin, valproate and vice versa. Erythromycin, fluoxetine, isoniazid inhibit metabolism of carbamazepine.
Uses
Carbamazepine is the most effective drug for CPS and shares first choice drug status with phenytoin for GTCS and SPS.
Trigeminal and related neuralgias: Carbamazepine is the drug of choice. These neuralgias are characterized by attacks of high intensity electric shocklike or stabbing pain set off by even trivial stimulation of certain trigger zones in the mouth or on the face. Drugs benefit by interrupting temporal summation of afferent impulses (by a selective action on high frequency nerve impulses). Carbamazepine is not an analgesic, but has a specific action (almost diagnostic) in these neuralgias. About 60% patients respond well. Phenytoin and baclofen are less efficacious alternatives.
Manic depressive illness and acute mania: as an alternative to lithium (see Ch. No. 32).
Dose: 200–400 mg TDS; Children 15–30 mg/kg/day. TEGRETOL, MAZETOL 100, 200, 400 mg tab, 100 mg/
5 ml syr; CARBATOL 100, 200, 400 mg tab. MAZETOL SR, TEGRITAL CR 200, 400 mg sustained release/continuous release tabs. to avoid high peaks and low troughs in plasma concentration.
Oxcarbazepine
This newer congener of carbamazepine is rapidly converted to an active metabolite that is only glucuronide conjugated but not oxidized. Toxic effects due to the epoxide metabolite are avoided. Drug interactions and autoinduction of own metabolism are less marked, because it is a weak enzyme inducer. Risk of hepatotoxicity is estimated to be lower than carbamazepine; but that of hyponatraemia is more. Indications are the same as for carbamazepine, but it may be better tolerated. Dose to dose it is 1½ times less potent.
OXETOL, OXCARB, OXEP 150, 300, 600 mg tabs.
Ethosuximide
The most prominent action of ethosuximide is antagonism of PTZ induced clonic seizures at doses which produce no other discernable action. It raises seizure threshold but does not modify maximal electroshock seizures or inhibit kindling. Clinically it is effective only in absence seizures.
The primary action appears to be exerted on thalamocortical system which is involved in the generation of absence seizures. The EEG in absence seizures shows characteristic bilaterally synchronous 3 Hz spike and wave rhythm generated by oscillation of impulses between thalamus and neocortex through reverberatory synaptic connections. Thalamic neurones exhibit prominent ‘T’ (transient) current which is low threshold Ca2+ current (due to inward flow of Ca2+ through T type Ca2+ channels) that acts as the pacemaker and amplifies repetitive spikes. Ethosuximide selectively suppresses T current without affecting other types of Ca2+ or Na+ currents. It also does not potentiate GABA at therapeutic concentrations. This correlates well with its selective action in absence seizures.
Ethosuximide is rather slowly but completely absorbed, not protein bound, evenly distributed in body, and largely metabolized in liver by hydroxylation and glucuronidation, and excreted in urine—about ¼th in the unchanged form. Plasma t½ averages 48 hours in adults and 32 hours in children.
Adverse Effects
Doserelated side effects are gastrointestinal intolerance, tiredness, mood changes, agitation, headache, drowsiness and inability to concentrate.
Hypersensitivity reactions like rashes, DLE and blood dyscrasias are rare. No liver or kidney damage.
Use
The only indication for ethosuximide is absence seizures; in that also it has been superseded by valproate. Dose: 20–30 mg/kg/day; ZARONTIN 250 mg/5 ml syr.
Valproic Acid (Sodium Valproate)
It is a branched chain aliphatic carboxylic acid with a broad spectrum anticonvulsant action. It is more potent in blocking PTZ seizures than in modifying maximal electroshock. Establishment of chronic experimental seizure foci and kindling are also prevented. Remarkably, at anticonvulsant doses, valproate produces little sedation or other central effects. Likewise, it is effective in partial seizures and GTCS as well as absence seizures.
Valproate appears to act by multiple mechanisms:
• A phenytoin-like frequency-dependent prolongation of Na+ channel inactivation.
• Weak attenuation of Ca2+ mediated ‘T’ current (ethosuximide like).
• Augmentation of release of inhibitory transmitter GABA by inhibiting its degradation (by GABA-transaminase) as well as probably by increasing its synthesis from glutamic acid. However, responses to exogenously applied GABA are not altered.
Pharmacokinetics
Oral absorption of valproic acid is good. It is 90% bound to plasma proteins; completely metabolized in liver by oxidation (some metabolites are active) and glucuronide conjugation—excreted in urine. Plasma t½ is 10– 15 hours; but anticonvulsant effects are longer lasting.
Adverse Effects
The toxicity of valproate is low.
Anorexia, vomiting, heart burn are common. Drowsiness, ataxia and tremor are doserelated side effects. However, cognitive and behavioral effects are not prominent.
Alopecia, curling of hair and increased bleeding tendency have been observed.
Rashes and thrombocytopenia are infrequent hypersensitivity phenomena.
Asymptomatic rise in serum transaminase is common; monitoring of liver function is advised. A rare but serious adverse effect is fulminant hepatitis; occurs only in children (especially below 3 yr). Those with hepatic disease or who receive other anticonvulsant or hepatotoxic drug are at greater risk. Pancreatitis is also reported. Long-term use of valproate in young girls has been associated with higher incidence of polycystic ovarian disease and menstrual irregularities.
Used during pregnancy, it has produced spina bifida and other neural tube defects in the offspring; should be avoided.
Dose: Adults—start with 200 mg TDS, maximum 800 mg TDS; children—15–30 mg/kg/day.
VALPARIN CHRONO 200, 300, 500 mg tabs, 200 mg/ 5 ml syr; ENCORATE 200, 300, 500 mg regular and controlled release tabs, 200 mg/5 ml syr, 100 mg/ml inj.
Uses
Valproic acid is the drug of choice for absence seizures.
It is an alternative/adjuvant drug for GTCS, SPS and CPS.
Myoclonic and atonic seizures—control is often incomplete, but valproate is the drug of choice. Mania and bipolar illness: as alternative to lithium.
Valproate has some prophylatic efficacy in migraine.
Interactions
• Valproate increases plasma levels of phenobarbitone by inhibiting its metabolism.
• It displaces phenytoin from protein binding site and decreases its metabolism → phenytoin toxicity.
• Valproate and carbamazepine induce each other’s metabolism.
• Concurrent administration of clonazepam and valproate is contraindicated because absence status may be precipitated.
• Foetal abnormalities are more common if valproate and carbamazepine are given concurrently.
Divalproex (Semisodium valproate)
It is the coordination compound of valproic acid with sodium valproate (1:1). Oral absorption is slower, but bioavailability is the same. Gastric tolerance may be better.
DIPROEX, VALANCE, 125, 250, 500 mg tabs; DEPAKOTE 250, 500 mg tabs.
Clonazepam
It is a benzodiazepine with prominent anticonvulsant properties: blocks PTZ seizures at doses which produce mild sedation. Efficacy in modifying maximal electroshock seizures is low. Though in experimental models of chronic epilepsy it inhibits spread rather than the focus itself, it is singularly ineffective in GTCS.
Production of generalized seizures by kindling is suppressed, but local after-discharges persist.
Benzodiazepines potentiate GABA induced Cl– influx to produce sedation and the same mechanism has been held responsible for the anticonvulsant property, but the sites of action in the brain may be different. At large doses, high frequency discharges are inhibited akin to phenytoin.
Pharmacokinetics
Oral absorption of clonazepam is good. It is 85% bound to plasma proteins, completely metabolized in liver and excreted in urine; t½ averages 24 hours. It does not produce any active metabolite.
Adverse Effects
The most important side effect of clonazepam is sedation and dullness. This can be minimized by starting at low dose; some tolerance develops with chronic therapy. Lack of concentration, irritability, temper and other behavioral abnormalities may occur in children. Motor disturbances and ataxia are doserelated adverse effects.
Salivation and increased respiratory secretions may be complained of.
Uses
Clonazepam has been primarily employed in absence seizures. It is also useful as an adjuvant in myoclonic and akinetic epilepsy and may afford some benefit in infantile spasms. However, its value is limited by development of tolerance to the therapeutic effect within six months or so.
Dose: adults 0.5–5 mg TDS, children 0.02–0.2 mg/kg/ day.
LONAZEP, CLONAPAX, RIVOTRIL 0.5, 1.0, 2.0 mg tab.
Clobazam
It is a 1,5 benzodiazepine (diazepam and others are 1,4 benzodiazepines) introduced first as anxiolytic and later found to possess useful antiepileptic efficacy in partial, secondarily generalized tonic-clonic as well as absence, myoclonic and atonic seizures, including some refractory cases. Sedation and psychomotor retardation are less prominent, but side effect profile is similar to other BZDs. It appears to act by facilitating GABA action.
Oral bioavailability of clobazam is ~90% and elimination t½ 18 hrs, but an active metabolite is produced which has longer t½ (>35 hr). It is generally used as adjuvant to other antiepileptic drugs like phenytoin, carbamazepine, valproate or phenobarbitone in refractory epilepsy. The above drugs may lower serum levels of clobazam.
Dose: start with 10–20 mg at bedtime, can be increased upto 60 mg/day; FRISIUM, LOBAZAM, CLOZAM, 5, 10, 20 mg cap.
Diazepam (see Ch. No. 29)
It has anticonvulsant activity in a variety of models but is not used for long term therapy of epilepsy because of prominent sedative action and rapid development of tolerance to the antiepileptic effect. However, it is the drug of choice for emergency control of convulsions, e.g. status epilepticus, tetanus, eclampsia, convulsant drug poisoning, etc.
For this purpose 0.2–0.5 mg/kg slow i.v. injection is followed by repeated doses as required; maximum 100 mg/day. Thromboflebitis of injected vein is not uncommon. Marked fall in BP and respiratory depression can occur; resuscitative measures should be at hand before the drug is injected.
Rectal instillation of diazepam is now the preferred therapy for febrile convulsions in children.
Lorazepam 0.1 mg/kg injected i.v. at a rate not exceeding 2 mg/min is an alternative to diazepam in status epilepticus or for emergency control of convulsions of other etiology. The action of lorazepam after i.v. injection is more sustained than that of diazepam which is rapidly redistributed.
Lamotrigine
A new anticonvulsant having carbamazepine-like action profile: modifies maximal electroshock and decreases electrically evoked as well as photic after discharge duration. Prolongation of Na+ channel inactivation and suppression of high frequency firing has been demonstrated. In addition, it may directly block voltage sensitive Na+ channels, thus stabilizing the presynaptic membrane and preventing release of excitatory neurotransmitters, mainly glutamate and aspartate. This may account for its broader-spectrum of antiseizure efficacy. However, it does not antagonize PTZ seizures or block NMDA type of glutamate receptors.
Lamotrigine is a broad-spectrum antiepileptic. Initially found useful as addon therapy in refractory cases of partial seizures and GTCS, it has now been shown effective as monotherapy as well. Absence and myoclonic or akinetic epilepsy cases have also been successfully treated. Reduction in seizure frequency or complete control is obtained as frequently as with carbamazepine.
Lamotrigine is well absorbed orally and metabolized completely in liver. Its t½ is 24 hr, but is reduced to ~16 hr in patients receiving phenytoin, carbamazepine or phenobarbitone. On the contrary valproate inhibits glucuronidation of lamotrigine and doubles its blood level, but valproate levels are lowered by lamotrigine. Reduce the dose of lamotrigine to half in patients taking valproate. However, metabolism of other anticonvulsants and oral contraceptives is not altered.
Side effects are sleepiness, dizziness, diplopia, ataxia and vomiting. In some comparative trials lamotrigine has been found to be better tolerated than carbamazepine or phenytoin. Negative effect on cognitive function is not reported. Rash may be a severe reaction, particularly in children, requiring withdrawal.
Dose: 50 mg/day initially, increase upto 300 mg/day as needed; not to be used in children. LAMETEC, LAMITOR, LAMIDUS 25, 50, 100 mg tabs.
Gabapentin
This lipophilic GABA derivative crosses to the brain and enhances GABA release, but does not act as agonist at GABAA receptor. It modifies maximal electroshock as well as inhibits PTZ induced clonic seizures. Added to a first line drug, it reduces seizure frequency in refractory partial seizures with or without generalization. Though gabapentin has been found effective as monotherapy as well in SPS and CPS, it is mostly employed as addon drug. Gabapentin is considered to be a first line drug for pain due to diabetic neuropathy and postherpetic neuralgia; has some prophylactic effect in migraine also.
Gabapentin is well absorbed orally and excreted unchanged in urine with a t½ of 6 hrs.
No change in dose of primary antiepileptic drug is required when gabapentin is added. Side effects are mild sedation, tiredness, dizziness and unsteadiness.
Dose: Start with 300 mg OD, increase to 300–600 mg
TDS as required; NEURONTIN 300 mg, 400 mg cap, GABANTIN, GABAPIN 100, 300, 400 mg cap.
Vigabatrin (γ vinyl GABA)
It is an inhibitor of GABA-transaminase, the enzyme which degrades GABA. Anticonvulsant action may be due to increase in synaptic GABA concentration. It suppresses maximal electroshock and kindled seizures, and is effective in many patients with refractory epilepsy, especially partial seizures with or without generalization. It is at present approved only for adjuvant medication.
Visual field contraction and production of behavioral changes, depression and psychosis in some patients is its most important drawback.
Dose: 2–4 g daily; children 40–100 mg/kg/day.
Topiramate
This weak carbonic anhydrase inhibitor has broad spectrum anticonvulsant activity in maximal electroshock, PTZ induced clonic seizures and in kindling model. It appears to act by multiple mechanisms, viz phenytoin like prolongation of Na+ channel inactivation, GABA potentiation by a postsynaptic effect and antagonism of certain glutamate receptors.
Topiramate is indicated for supplementing primary antiepileptic drug in refractory SPS, CPS and GTCS. As monotherapy also its efficacy has been rated equivalent to valproate. Promising results have been obtained in myoclonic epilepsy. Topiramate is readily absorbed orally and primarily excreted unchanged in urine with an average t½ of 24 hours. Adverse effects are impairment of attention, sedation, ataxia, word finding difficulties, psychiatric symptoms, weight loss, paresthesias and renal stones.
Recently, topiramate has been approved for prophylaxis of migraine; may be used when β blockers/other prophylactics are contraindicated or are not effective.
Dose: Initially 25 mg OD, increase weekly upto 100–200 mg BD as required.
TOPEX, EPITOP, TOPAMATE, 25, 50, 100 mg tabs.
Tiagabine
This newer anticonvulsant potentiates GABA mediated neuronal inhibition by depressing GABA transporter GAT1 which removes synaptically released GABA into neurones and glial cells. Maximal electroshock and kindled seizures are suppressed. Currently it is approved only for addon therapy of partial seizures with or without secondary generalization, when not adequately controlled by standard antiepileptic drugs alone. Side effects are mild sedation, nervousness, asthenia, amnesia and abdominal pain.
Zonisamide
Another new anticonvulsant with weak carbonic anhydrase inhibitory action that modifies maximal electroshock seizures and inhibits kindling, but does not antagonize PTZ. Prolongation of Na+ channel inactivation resulting in suppression of repetitive neuronal firing has been observed. It is indicated as addon drug in refractory partial seizures.
Levetiracetam
A unique anticonvulsant which suppresses kindled seizures but is ineffective against maximal electroshock or PTZ. Clinical efficacy has been shown as adjuvant medication in refractory partial seizures with or without secondary generalization. None of the usual anticonvulsant mechanisms of action appear to be applicable to levetiracetam.
