Antitubercular Drugs
| Home | | Pharmacology |Chapter: Essential pharmacology : Antitubercular Drugs
Tuberculosis is a chronic granulomatous disease and a major health problem in developing countries. About 1/3rd of the world’s population is infected with Mycobact. tuberculosis. As per WHO estimate, 9 million people globally develop active TB and 1.7 million die of it annually. In India, it is estimated that nearly 2 million people develop active disease every year and about 0.5 million die from it.
ANTITUBERCULAR DRUGS
Tuberculosis is a
chronic granulomatous disease and a major health problem in developing countries.
About 1/3rd of the world’s population is infected with Mycobact. tuberculosis. As per WHO estimate, 9 million people
globally develop active TB and 1.7 million die of it annually. In India, it is
estimated that nearly 2 million people develop active disease every year and
about 0.5 million die from it.
A new dimension got
added in the 1980s due to spread of HIV with high prevalence of tuberculosis
and Mycobact. avium complex (MAC)
infection among these patients. India has a large load of HIV infected subjects,
and these patients are especially vulnerable to severe forms of tubercular/MAC
infection. While lately, the increase in TB case rate associated with HIV
infection has been halted in the USA, no such trend is apparent in India. Emergence
of ‘multidrug resistant’ (MDR) TB of which over 0.4 million cases are occurring
globally every year, is threatening the whole future of current antitubercular
chemotherapy.
Remarkable progress
has been made in the last 60 years since the introduction of Streptomycin in 1947 for the treatment
of tuberculosis. Its full therapeutic potential could be utilized only after
1952 when isoniazid was produced to accompany
it. The discovery of ethambutol in
1961, rifampin in 1962, and redefinition
of the role of pyrazinamide has changed
the strategies in the chemotherapy of
tuberculosis. Since 1970 the efficacy of short course (6–9 months) and domiciliary
regimens has been demonstrated and clearcut treatment guidelines have been
formulated.
Fluoroquinolones,
newer macrolides and some rifampin congeners are the recent additions to the
antimycobacterial drugs. According to their clinical utility the antiTB drugs
can be divided into:
First line: These drugs have high antitubercular efficacy as well as low toxicity; are used
routinely.
Second line: These drugs have either low antitubercular efficacy or high toxicity or
both; are used in special circumstances only.
First Line Drugs
1. Isoniazid (H)
2. Rifampin (R)
3. Pyrazinamide (Z)
4. Ethambutol (E)
5. Streptomycin (S)
Second Line Drugs
1. Thiacetazone (Tzn)
2. Paraaminosalicylic acid (PAS)
3. Ethionamide (Etm)
4. Cycloserine (Cys)
5. Kanamycin (Kmc)
6. Amikacin (Am)
7. Capreomycin (Cpr)
Newer
Drugs
1. Ciprofloxacin
2. Ofloxacin
3. Clarithromycin
4. Azithromycin
5. Rifabutin
Isoniazid (Isonicotinic acid hydrazide, H)
Isoniazid is the antitubercular drug parexcellence, and an
essential component of all antitubercular regimens, unless the patient is not
able to tolerate it or bacilli are resistant. It is primarily tuberculocidal.
Fast multiplying organisms are rapidly killed, but quiescent ones are only
inhibited. It acts on extracellular as well as on intracellular TB (bacilli
present within macrophages); is equally active in acidic and alkaline medium.
It is one of the cheapest antitubercular drugs. However, most atypical
mycobacteria are not inhibited by INH.
The most plausible
mechanism of action of INH is inhibition of synthesis of mycolic acids which are unique fatty acid component of
mycobacterial cell wall. This may explain the high selectivity of INH for
mycobacteria (it is not active against any other microorganism). The lipid
content of mycobacteria exposed to INH is reduced. A gene labelled inh A which encodes for a fatty acid synthase
enzyme is the target of INH action. The sensitive mycobacteria concentrate INH
and convert it by a catalase-peroxidase enzyme into an active metabolite that interacts
with the inh A gene.
About 1 in 106
tubercle bacilli is inherently resistant to clinically attained INH concentrations.
If INH is given alone, such bacilli proliferate selectively and after 2–3
months (sometimes even earlier) an apparently resistant infection emerges. The
most common mechanism of INH resistance is by mutation of the catalase-peroxidase
gene so that the bacilli do not generate the active metabolite of INH. However,
bacilli that lose catalase activity also appear to become less virulent; many
physicians like to continue INH even when bacilli are apparently resistant to
it in vitro. INH resistance may also involve mutation in the target inh A
gene. Other resistant TB bacilli lose the active INH concentrating process. The
incidence of primary INH resistance varies widely (1–33%) among different
populations, depending on the extent of use and misuse of INH in that area.
Combined with other drugs, INH has good resistance preventing action. No cross
resistance with other antitubercular drugs occurs.
Pharmacokinetics
INH is completely
absorbed orally and penetrates
all body tissues, tubercular cavities, placenta and meninges. It is extensively
metabolized in liver; most important pathway being acetylation—metabolites are
excreted in urine. The rate of INH acetylation shows genetic variation. There
are either:
Fast acetylators
(30–40% of Indians) t½ of INH 1 hr.
Slow acetylators
(60–70% of Indians) t½ of INH 3 hr.
The proportion of fast
and slow acetylators differs in different parts of the world. However,
acetylator status does not matter if INH is taken daily, but biweekly regimens
are less effective in fast acetylators. Isoniazid induced peripheral neuritis
appears to be more common in slow acetylators.
Interactions Aluminium hydroxide inhibits INH absorption.
INH inhibits
phenytoin, carbamazepine, diazepam and warfarin metabolism: may raise their blood
levels.
PAS inhibits INH
metabolism and prolongs its t½.
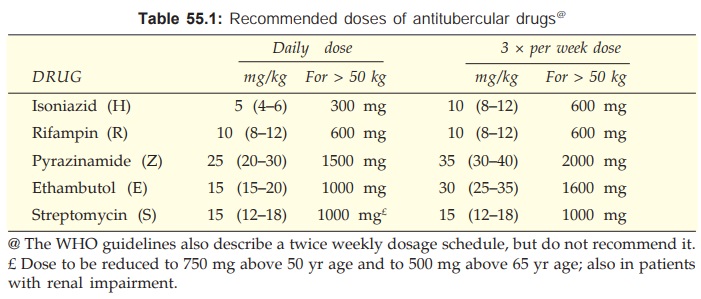
Dose of all first line drugs is given in Table 55.1.
Adverse Effects INH is well tolerated
by most patients. Peripheral
neuritis and a variety of neurological manifestations (paresthesias, numbness, mental
disturbances, rarely convulsions) are the most important dose-dependent toxic
effects. These are due to interference with utilization of pyridoxine and its
increased excretion in urine (see Ch.
No. 67). Pyridoxine given prophylactically (10 mg/day) prevents the
neurotoxicity even with higher doses, but routine use is not mandatory. INH
neurotoxicity is treated by pyridoxine 100 mg/day.
Hepatitis, a major adverse effect of INH, is rare in children,
but more common in older people and in alcoholics. It is due to dose-related
damage to liver cells and is reversible on stopping the drug.
Other side effects are rashes, fever, acne and arthralgia.
ISONEX 100, 300 mg tabs, ISOKIN 100 mg tab, 100 mg per 5 ml liq.
Rifampin (Rifampicin, R)
It is a semisynthetic derivative
of rifamycin B obtained from Streptomyces
mediterranei. Rifampin is bactericidal to M. tuberculosis and many other gram-positive and gram-negative
bacteria like Staph. aureus, N. meningitidis, H. influenzae, E.coli, Klebsiella, Pseudomonas, Proteus and
Legionella. Against TB bacilli, it is as efficacious as INH and better than all other drugs. The bactericidal action covers
all subpopulations of TB bacilli, but acts best on slowly or intermittently
(spurters) dividing ones, as well as on many atypical mycobacteria. Both extra
and intracellular organisms are affected. It has good sterilizing and
resistance preventing actions.
Rifampin inhibits DNA dependent RNA synthesis. Probably, the
basis of selective toxicity is that mammalian RNA polymerase does not avidly
bind rifampin.
Mycobacteria and other
organisms develop resistance to rifampin rather rapidly. However, the incidence
of resistant TB is less than 10–7 and it is quite unusual for a patient to have
primary rifampin resistant tubercular infection. Rifampin resistance is nearly
always due to mutation in the repoB
gene (for the β subunit of RNA polymerase—the
target of rifampin action) reducing its affinity for the drug. No cross
resistance with any other antitubercular drug has been noted.
Pharmacokinetics
It is well absorbed
orally, widely distributed in
the body: penetrates cavities, caseous masses, placenta and meninges. It is
metabolized in liver to an active deacetylated metabolite which is excreted
mainly in bile, some in urine also. Rifampin and its desacetyl derivative
undergo enterohepatic circulation. The t½ of rifampin is variable (2–5 hours).
Interactions
Rifampin is a
microsomal enzyme inducer—increases
several CYP450 isoenzymes, including CYP3A4, CYP2D6, CYP1A2 and CYP2C
subfamily. It thus enhances its own metabolism as well as that of many drugs
including warfarin, oral contraceptives, corticosteroids, sulfonylureas,
digitoxin, steroids, HIV protease inhibitors, nonnucleoside reverse
transcriptase inhibitors (NNRTIs), theophylline, metoprolol, fluconazole,
ketoconazole, etc. Contraceptive failures have occurred: switch over to an oral
contraceptive containing higher dose (50 μg) of estrogen or use
alternative method of contraception.
Adverse Effects
The incidence of
adverse effects is similar to INH.
Hepatitis, a major
adverse effect, generally occurs in patients with preexisting liver disease and
is doserelated: development of jaundice requires discontinuation of the
drug—then it is reversible. Other serious but rare reactions are:
· ‘Respiratory syndrome’: breathlessness which
may be associated with shock and collapse.
·
Purpura, haemolysis, shock and renal failure.
Minor reactions usually not requiring drug withdrawal and more common with
intermittent regimens are:
·
‘Cutaneous syndrome’: flushing, pruritus +
rash (especially on face and scalp), redness and watering of eyes.
·
‘Flu syndrome’: with chills, fever, headache,
malaise and bone pain.
· ‘Abdominal syndrome’: nausea, vomiting,
abdominal cramps with or without diarrhoea.
Urine and secretions may become orange-red— but this is
harmless.
Other Uses Of Rifampin
·
Leprosy (see
Ch. No. 56)
· Prophylaxis of Meningococcal and H. influenzae meningitis and carrier state.
· Second/third choice drug for MRSA,
diphtheroids and Legionella
infections.
· Combination of doxycycline and rifampin is the
first line therapy of brucellosis.
RCIN 150, 300, 450,
600 mg caps, 100 mg/5 ml susp. RIMACTANE, RIMPIN 150, 300, 450 mg caps, 100 mg/
5 ml syr.; RIFAMYCIN 450 mg cap, ZUCOX 300, 450, 600 mg tabs; to be taken 1
hour before or 2 hour after meals.
Pyrazinamide (Z)
Chemically similar to INH, pyrazinamide (Z) was developed parallel
to it in 1952. It is weakly tuberculocidal but more active in acidic medium. It
is more lethal to intracellularly located bacilli and to those at sites showing
an inflammatory response (pH is acidic at both these locations). It is highly
effective during the first 2 months of therapy when inflammatory changes are
present. By killing the residual intracellular bacilli it has good
‘sterilizing’ activity. Its use has enabled regimens to be shortened and risk
of relapse to be reduced. Mechanism of antimycobacterial action of Z resembles
INH; it inhibits mycolic acid synthesis, but by interacting with a different
fatty acid synthase encoding gene. Resistance to Z develops rapidly if it is
used alone, and is due to mutation in the pncA
gene which encodes for the enzyme generating the active metabolite of Z.
Pyrazinamide is absorbed orally, widely distributed, has good
penetration in CSF, extensively metabolized in liver and excreted in urine;
plasma t½ is 6–10 hours.
Hepatotoxicity is the most important doserelated adverse effect,
but it appears to be less
common in the Indian
population than in western countries. Daily dose is now limited to 25–30 mg/ kg
which produces only a low incidence of hepatotoxicity. It is contraindicated in
patients with liver disease.
Hyperuricaemia is common
and is due to inhibition of uric acid secretion in kidney: gout can occur.
Other adverse effects
are arthralgia, flushing, rashes, fever and loss of diabetes control.
PYZINA 0.5, 0.75, 1.0
g tabs, 0.3 g kid tab; PZACIBA 0.5, 0.75 g tabs, 250 mg/5 ml syr; RIZAP 0.75,
1.0 g tabs.
Ethambutol (E)
Ethambutol is
selectively tuberculostatic and clinically as active as S. Fast multiplying
bacilli are more susceptible as are many atypical mycobacteria. Added to the
triple drug regimen of RHZ it has been found to hasten the rate of sputum
conversion and to prevent development of resistance.
The mechanism of
action of E is not fully understood, but it has been found to inhibit
arabinosyl transferases involved in arabinogalactan synthesis and to interfere
with mycolic acid incorporation in mycobacterial cell wall. Resistance to E
develops slowly; in many cases it is due to alteration in the drug target gene.
No cross resistance with any other antitubercular drug has been noted.
About 3/4 of an oral
dose of E is absorbed. It is distributed widely but penetrates meninges
incompletely and is temporarily stored in RBCs. Less than ½ of E is
metabolized. It is excreted in urine by glomerular filtration and tubular
secretion; plasma t½ is ~4 hrs. Caution is required in its use in patients with
renal disease.
Patient acceptability
of E is very good and side effects are few. Loss of visual acuity/colour vision,
field defects due to optic neuritis is the most important dose and duration of
therapy dependent toxicity. Because young children may be unable to report
early visual impairment, it should not be used below 6 years of age. With early
recognition and stoppage of therapy, visual
toxicity is largely reversible. Ethambutol produces few other
symptoms: nausea, rashes, fever, neurological changes are infrequent.
Hyperuricemia is due to interference with urate excretion. It is a commonly
used antitubercular drug.
MYCOBUTOL, MYAMBUTOL, COMBUTOL 0.2, 0.4, 0.6, 0.8, 1.0 g tabs.
Streptomycin (S)
The pharmacology of streptomycin is described in Ch. No. 53. It
was the first clinically useful antitubercular drug. It is tuberculocidal, but
less effective than INH or rifampin; acts only on extracellular bacilli
(because of poor penetration into cells). Thus, host defence mechanisms are
needed to eradicate the disease. It penetrates tubercular cavities, but does
not cross to the CSF, and has poor action in acidic medium.
Resistance developed
rapidly when streptomycin was used alone in tuberculosis—most patients had a
relapse. In an average population of TB, 1 in 108 to 1 in 106 bacillus is
resistant to S; these bacilli selectively multiply and stage a comeback after
initial control. In case of S-resistant infection, it must be stopped at the
earliest because of chances of S-dependence—the infection flourishing when the
drug is continued. Most atypical mycobacteria are unaffected by S.
Popularity of S in the treatment of tuberculosis had declined
due to need for i.m. injections and lower margin of safety, because of ototoxicity
and nephrotoxicity, especially in the elderly and in those with impaired renal
function.
Thiacetazone
(Tzn, Amithiozone)
Thiosemicarbazones were the first antitubercular drugs tested,
but were weak. Domagk studied their action. Thiacetazone was found to be the best
out of many derivatives. It was tried in the west, found to be hepatotoxic and
discarded. In India, interest in Tzn was revived in the 1960s for oral use
along with INH as a substitute for PAS. Though, its importance has declined, it
continues to be used as a convenient low cost drug to prevent emergence of
resistance to INH and more active agents.
Thiacetazone is a tuberculostatic, low efficacy drug; does not
add to the therapeutic effect of H, S or E, but delays resistance to these drugs.
It is orally active, and primarily excreted unchanged in urine with a t½ of 12
hr.
The major adverse
effects of Tzn are hepatitis, exfoliative dermatitis, Stevens-Johnson syndrome
and rarely bone marrow depression. The common side effects are anorexia,
abdominal discomfort, loose motions and minor rashes. A mild anaemia persists
till Tzn is given. Tzn is a reserve anti-TB drug, sometimes added to INH in
alternative regimens. It should not be used in HIV positive cases, because incidence
of serious toxicity is higher.
Dose: 150 mg OD in adults,
2.5 mg/kg in children. It is frequently
used as combined tablet with isoniazid.
Paraamino salicylic acid (PAS)
Introduced in 1946, it
is related to sulfonamides— chemically as well as in mechanism of action. It is
not active against other bacteria: selectivity may be due to difference in the
affinity of folate synthase of TB and other bacteria for PAS.
PAS is tuberculostatic
and one of the least active drugs: does not add to the efficacy of more active
drugs that are given with it; only delays development of resistance— probably,
by directly inhibiting episomal resistance transfer. Resistance to PAS is slow
to develop. It is used as the sodium salt (large doses that are needed may
cause Na+ overload) or calcium salt (better gastric tolerance is claimed).
PAS is absorbed
completely by the oral route and distributed all over except in CSF. About 50%
PAS is acetylated; competes with acetylation of INH—prolongs its t½. PAS
formulations interfere with absorption of rifampin. It is excreted rapidly by
glomerular filtration and tubular secretion; t½ is short, ~1 hour.
Patient acceptability
of PAS is poor because of frequent anorexia, nausea and epigastric pain. Other
adverse effects are rashes, fever, malaise, goiter, liver dysfunction and blood
dyscrasias.
Dose: 10–12 g (200 mg/kg)
per day in divided doses; SODIUMPAS 0.5 g tab,
80 g/100 g granules. It is rarely used now.
Ethionamide (Etm)
It is a
tuberculostatic drug of moderate efficacy introduced in 1956. It acts on both extra
and intracellular organisms. Atypical mycobacteria are sensitive. Resistance to
Etm develops rapidly and some cross resistance with Tzn is seen. It is absorbed
orally, distributes all over, including CSF, completely metabolized and has a short
duration of action (t½ 2–3 hr).
Anorexia, nausea,
vomiting and abdominal upset are common, especially in Indian patients. Though
the recommended dose of Etm is 1 g/day, more than 0.5 g is generally not
tolerated. Other side effects are aches and pains, rashes, hepatitis,
peripheral or optic neuritis, mental disturbances and impotence. It is seldom
used; only in case of resistance to better tolerated drugs.
Dose: 0.5–0.75 g (10–15
mg/kg) per day; ETHIDE, ETHIOCID,
MYOBID 250 mg tab.
Cycloserine (Cys)
It is an antibiotic
obtained from S. orchidaceus, and is
a chemical analogue of Dalanine: inhibits bacterial cell wall synthesis by
inactivating the enzymes which recemize Lalanine and link two D alanine
residues. Cys is tuberculostatic and inhibits some other gram-positive bacteria,
E. coli, Chlamydia also. Resistance to Cys develops slowly: no cross
resistance.
Cycloserine is absorbed orally, diffuses all over, CSF
concentration is equal to that in plasma. About 1/3rd of a dose is metabolized,
the rest is excreted unchanged by kidney. The CNS toxicity of Cys is
high—sleepiness, headache, tremor and psychosis; convulsions may be prevented
by pyridoxine 100 mg/day. It is rarely used; only in resistant cases. The shelf
life of Cys in warm climate is short.
Dose: 250 mg BD, increased
if tolerated upto 500 mg BD.
CYCLORINE, COXERIN,
MYSER 250 mg cap.
Kanamycin, Amikacin and Capreomycin are more toxic antibiotics used as reserve drugs in
rare cases not responding to the usual therapy, or infection by atypical
mycobacteria. Any one of these is used at a time in combination with the
commonly employed drugs to which resistance has not developed. Because all
exhibit similar ototoxicity and nephrotoxicity, they are not combined among
themselves or with streptomycin. Capreomycin, in addition, can induce electrolyte
abnormalities. All act by inhibiting protein synthesis. None is effective
orally; none penetrates meninges. All are excreted unchanged by the kidney. All
are given in a dose of 0.75–1.0 g i.m. per day.
Kanamycin and amikacin
are aminoglycosides and have been described in Ch. No. 53. Amikacin is a very
promising drug for atypical mycobacteria including M. avium. Capreomycin is available as
KAPOCIN 0.5, 0.75, 1.0
g inj.
NEWER DRUGS
Ciprofloxacin, Ofloxacin, Moxifloxacin (see Ch 50 for description) The fluoroquinolones are a
useful new addition to the antitubercular drugs. Ciprofloxacin, ofloxacin,
moxifloxacin, gatifloxacin and sparfloxacin are active against M. tuberculosis
as well as M. avium complex (MAC) and M.
fortuitum. They penetrate cells and kill mycobacteria lodged in macrophages
as well. Because of their good tolerability, ciprofloxacin and ofloxacin are
being increasingly included in combination regimens against MDR tuberculosis
and MAC infection in HIV patients. They are also being used to supplement
ethambutol + streptomycin in cases when H, R, Z have been stopped due to
hepatotoxicity. However, neither ciprofloxacin nor ofloxacin have enhanced the
sterilizing ability of long-term regimens containing H and R. The generally
employed doses are ciprofloxacin 1500 mg/day and ofloxacin 800 mg/day in 2
divided doses. Sparfloxacin is more active against mycobacteria in vitro, but has been used clinically
to a lesser extent.
Clarithromycin, Azithromycin
These newer macrolide
antibiotics are most active against nontubercular mycobacteria including MAC, M. fortuitum, M. Kansasii and M. marinum.
Clarithromycin has been used to a greater extent because its MIC values are
lower, but azithromycin may be equally efficacious due to its higher tissue and
intracellular levels. For MAC and other atypical mycobacterial infection the
dose of clarithromycin is 500 mg BD and that of azithromycin 500 mg OD in
combination with other drugs. In AIDS patients, lifelong therapy is required—may
cause ototoxicity.
Rifabutin
It is related to
rifampin in structure and mechanism of action;
but less active against M. tuberculosis
and more active against MAC. Only partial cross resistance occurs between the
two. In a dose of 300 mg/day rifabutin is used for prophylaxis of MAC infection
in AIDS patients. For the treatment of established MAC infection, it has been
added to ethambutol + clarithromycin/azithromycin. Gastrointestinal intolerance,
rashes, granulo-cytopenia, myalgia and uveitis have been noted as adverse
effects. Reactions similar to those produced by rifampin can also occur. Like
rifampin, it is an enzyme inducer, but weaker. It is substituted for rifampin
for M. tuberculosis infection in HIV
patients who receive a protease inhibitor and/or a NNRTI, whose metabolism is
markedly induced by rifampin.
Some antitubercular combinations
RIFATER: Rifampin 120 mg,
isoniazid 80 mg, pyrazinamide 250 mg tab.
RCINEX: Rifampin 600
mg, isoniazid 300 mg tab; R
CINEXZ: Rifampin 225
mg, isoniazid 150 mg, pyrazinamide 750 mg tab.
RIMACTAZID, RIFADININH,
Rifampin 450 mg, isoniazid 300 mg tab.
MYCONEX 600 and 800;
Isoniazid 300 mg, ethambutol 600 mg or 800 mg tab, COMBUNEX Isoniazid 300 mg,
ethambutol 800 mg tab.
ARZIDE, ISORIFAM: Rifampin
450 mg, isoniazid 300 mg cap.
BITEBEN, ISOZONE,
UNITHIBEN: Isoniazid 75 mg, thiacetazone 37.5 mg tab, ISOZONE FORTE—double
strength.
INAPAS: sod PAS 834 mg, isoniazid 25 mg tab; sod PAS 3.34 g +
isoniazid 100 mg per measure granules. INABUTOL: Isoniazid 150 mg, ethambutol
400 mg tab; INABUTOL FORTE—double strength. ISOKIN–300: Isoniazid 300 mg, vit B6
10 mg tab.
IPCAZIDE: Isoniazid
100 mg, vit B6 5 mg per 5 ml liq.
Antitubercular combipacks (packs of 1 day’s dose)
AKT4: R 450 mg 1 cap + Z 750 mg 2 tab + E 800 mg H 300 mg 1
tab.
AKT3:R 450 mg 1 cap +
E 800 mg H 300 mg 1 tab.
CX5:R 450 mg 1 cap + Z
750 mg 2 tab + E 800 mg H 300 mg pyridoxine 10 mg 1 tab.
RIFACOMZ and RIMACTAZIDEZ:R
450 mg H 300 mg 1 tab. + Z 750 mg 2 tab.
RIFACOMEZ: R 450 mg H 300 mg 1 tab. + Z 750 mg
2 tab + E 800 mg 1 tab.
Fixed dose combination of antitubercular drugs with vitamins
(except INH + Vit B6) are banned in India.
