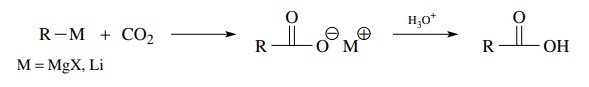Carboxylic Acids
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Functional Group Synthesis
Carboxylic acids have a relatively high oxidation level (+3), and thus a majority of synthetic methods to access carboxylic acids are oxidative in nature.
CARBOXYLIC ACIDS
Carboxylic
acids have a relatively high oxidation level (+3), and thus a majority of synthetic
methods to access carboxylic acids are oxidative in nature. Traditional
preparations include the following:
Oxidation
of olefins
Oxidation
of primary alcohols
Oxidation
of alkylbenzenes
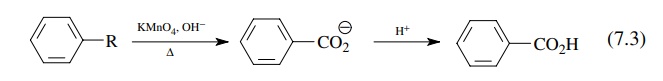
Hydrolysis
of nitriles
While
the above reactions will provide carboxylic acid products, each has problems
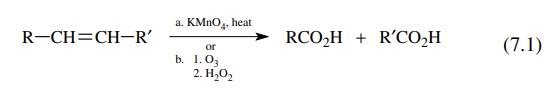
associated with it. The cleavage of olefins to carboxylic acids [reaction
(7.1)] can be carried out using potassium permanganate or by ozonolysis at low
temperature followed by oxidative workup with hydrogen peroxide. Neither of
these methods is very useful since only symmetric olefins provide a single
carboxylic acid product. Unsymmetrical olefins give a mixture of two acids
which must be separated. Furthermore the most useful synthetic processes are
those which build up structures, whereas these reactions are degradative in
nature.
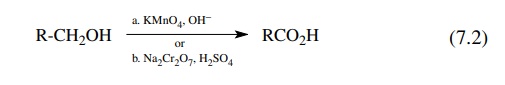
Primary
alcohols can be oxidized to carboxylic acids by a variety of reagents [reaction
(7.2)]. Often potassium permanganate or sodium dichromate were given as
reagents to use in this transformation. These are powerful oxidants, and many
other functional groups that might be present cannot survive the reaction
con-ditions. Milder oxidants are preferred and the best of these is chromic
acid in acetone (Jones reagent). Jones reagent is a mixture of chromic acid and
a stoi-chiometric amount of sulfuric acid which is needed in the redox process
to keep the solution at near a pH of 7. This technique is fast, easy, and
efficient and the reagent solution is easily prepared from chromium trioxide
and sulfuric acid in acetone. The oxidation can be carried out by adding the
Jones reagent by burette to the alcohol. Oxidation is instantaneous and the
addition can be stopped pre-cisely when all the alcohol has been consumed.
Using a stoichiometric amount of chromic acid usually leaves other functional
groups untouched. This is the method of choice for the synthesis of carboxylic
acids from primary alcohols.
The
oxidation of alkyl benzenes to benzoic acids [reaction (7.3)] is still carried
out occasionally, and this oxidation is most likely the only one where
potassium permanganate is the reagent of choice. Any carbon group attached to
the aromatic ring is degraded to the carboxylic acid group under the very
vigorous conditions of this oxidation.
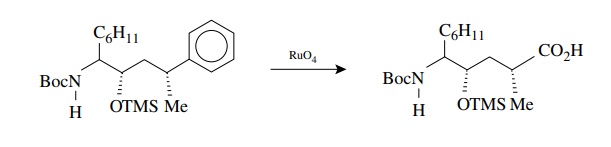
An
interesting twist on the oxidation of aromatic compounds forms the basis of a
new and very useful synthesis of carboxylic acids. Normally the aromatic ring
is resistant to oxidation and the side chains are oxidatively degraded to
car-boxylic acids, as in reaction (7.3). It has been found that ruthenium
tetroxide is a mild and selective oxidant of aromatic rings and completely
degrades the ring to the carboxylic acid but leaves aliphatic groups
unoxidized. This is essentially the reverse of the chemoselectivity seen in
potassium permanganate oxidations of arenes, where the side chains are oxidized
but the aromatic ring is left intact. The selectivity and mildness is seen in
the following example in which no amide or silyl ether (OTMS) oxidation was
observed and there was no epimerization of either chiral center:
Another
common way to install a carboxylic acid group is to hydrolyze a carboxylic acid
derivative. Such hydrolyses do not require a change in oxidation level
(carboxylic acid derivatives are at the oxidation level of the acid itself),
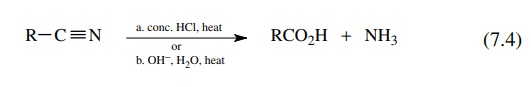
but they do normally require acid or base catalysis. Nitriles [reaction (7.4)]
often require vigorous conditions for hydrolysis mainly because they are only
weakly electrophilic. Either concentrated hydrochloric acid or sodium hydroxide
can be used to hydrolyze nitriles. The first stage of the hydrolysis produces
an amide and the amide is subsequently hydrolyzed to the acid. Each step of the
hydrolysis requires strenuous conditions and is useful mainly for nitriles that
lack other functional groups that would be destroyed by the stringent
conditions. It has been found that a mixture of sodium hydroxide and hydrogen
peroxide can be used to hydrolyze nitriles more efficiently than sodium
hydroxide alone and is the reagent of choice, although many other reagent
combinations have been reported.
Amides
are stabilized by resonance and are thus difficult to hydrolyze. Like nitriles
they can be hydrolyzed by concentrated hydrochloric acid or concentrated sodium
hydroxide. These are powerful reagents that tend to react with many other
functional groups as well. Thus hydrolysis is not a satisfactory method for
amides with many other functional groups present. As with nitriles, a mixture
of sodium hydroxide and hydrogen peroxide is one of the more effective reagents
for hydrolysis.
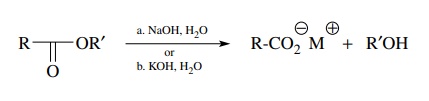
Esters,
on the other hand, are very common hydrolytic precursors to carboxylic acids.
The traditional reaction for the hydrolysis of esters is basic saponification
using sodium hydroxide or potassium hydroxide. While acid catalysis can also be
employed, preparative methods usually use base catalysis because formation of
the carboxylate salt drives the reaction to the right and gives high yields of
products.
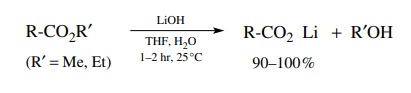
While
esters are much more easily hydrolyzed than amides, traditional saponi-fication
suffers from the fact that most esters are not soluble in aqueous base and so
the rate of the hydrolysis is limited by the solubility, not by the reactivity.
This limitation is overcome by the use of lithium hydroxide in aqueous THF, the
reagent of choice for basic hydrolysis of esters. Methyl and ethyl esters are
cleaved readily by this combination as most esters are soluble in this solvent mixture.
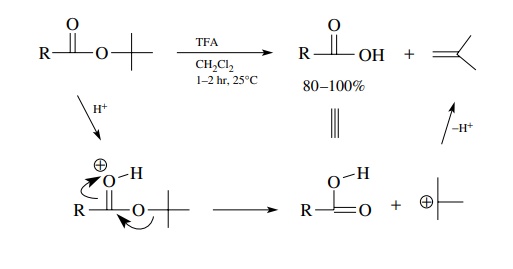
If
structural constraints prevent the use of basic hydrolysis of the ester group,
acid hydrolysis must be used. Nowadays it is much more common, in such
instances, to use tert-butyl esters
because they are cleaved rapidly and efficiently by trifluoroacetic acid (TFA)
to the carboxylic acid and isobutylene. This cleav-age is different from the
normal acid-catalyzed hydrolysis of esters in that the alkyl– oxygen bond is
broken rather than the acyl– oxygen bond. This change in mechanism is brought
about by the stability of the tert-butyl
cation which is produced upon alkyl– oxygen cleavage. As an added benefit, the
isobutylene by-product is a gas which escapes from the reaction mixture.
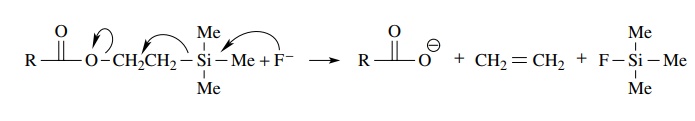
If
neither acidic nor basic conditions are compatible with other groups present in
the ester to be hydrolyzed, then β
-trimethylsilylethyl esters are often prepared. Trimethylsilylethyl esters are
cleaved easily by fluoride under mild, neutral con-ditions. Typical sources of
fluoride are cesium fluoride (CsF) or the more soluble tetrabutylammonium
fluoride (TBAF).
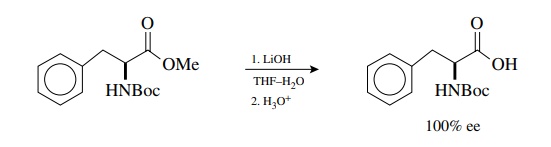
Current
methods for the hydrolysis of esters are fast, efficient, and sufficiently mild
that they are compatible with the presence of a variety of other func-tional
groups and/or stereocenters in the molecule. For example, protected amino acid
esters are hydrolyzed quantitatively without racemization or deprotection by
LiOH in aqueous THF.
Carboxylic
acid groups can also be installed in molecules using the reaction of an
organometallic compound with carbon dioxide. This is a reductive method since
the carbon dioxide is reduced to a carboxylic acid by formation of a new
car-bon – carbon bond. Both Grignard reagents and organolithium compounds work
well in this reaction.