Legal Basis, Principles and Organisation of the EU Pharmacovigilance System
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Regulatory Pharmacovigilance in the EU
The concept of pharmacovigilance was introduced into the legislation at EU level in 1993 through a Council Directive (Council Directive 93/39/EEC amending Council Directive 75/319/EEC).
LEGAL BASIS, PRINCIPLES AND
ORGANISATION OF THE EU PHARMACOVIGILANCE SYSTEM
The
concept of pharmacovigilance was introduced into the legislation at EU level in
1993 through a Council Directive (Council Directive 93/39/EEC amending Council
Directive 75/319/EEC). EU medicines legislation has since been codified into a
single Directive (2001/83/EC) in which pharma-covigilance is covered in Title
IX (Articles 101–108). Directives of the European Parliament and the Council
have the objective of harmonising the national legis-lation of the EU Member
States, and Member States are bound to implement these legal provisions into
their national legislations. However, pharmacovigi-lance systems already
existed in most countries which were Member States in 1993 and also in many of
those joining the EU through the enlargement process in 2004. These systems
vary according to differences in historical development and the organisation of
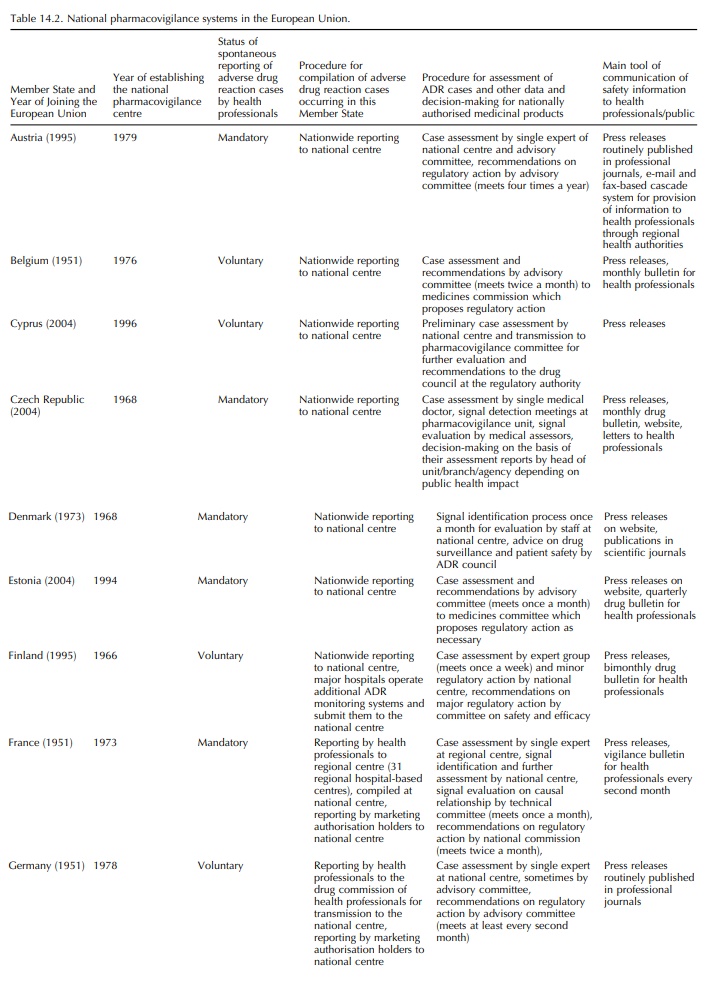
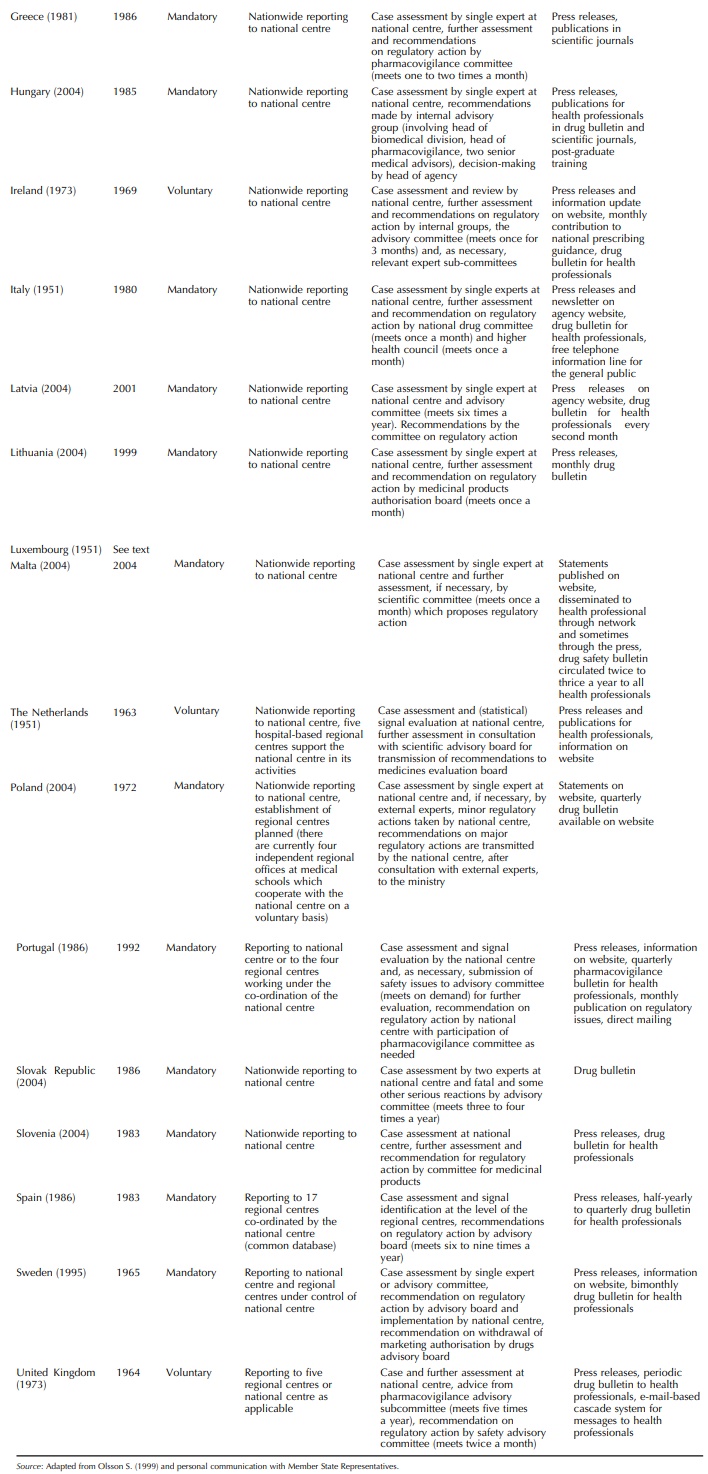
health-care at national level. Table 14.2 summarises the organisational
features of the national pharmacovigi-lance systems. All are an integral part
of the respective national drug regulatory authority (except in Luxem-bourg for
which spontaneous reports are submitted to one of the French regional centres
located in Nancy). Through the EU legislation, their activities are speci-fied
with regard to medicinal products authorised for use on their territory as
follows:
·
to collect information about suspected ADRs that occur under
normal conditions of use;
·
to obtain information on consumption data;
·
to collate information on misuse and abuse;
·
to evaluate this information scientifically; and
·
to ensure the adoption of appropriate regulatory decisions.
Practice
has shown that pharmacovigilance needs to be conducted with a view to how the
product is used in ordinary clinical practice. This includes use outside the
terms of the marketing authorisation. Experience gained during the
post-authorisation phase may also provide valuable input into the evaluation of
medici-nal products at the stage of application for marketing authorisation, if
there are chemical or pharmacologi-cal similarities with authorised products.
The national pharmacovigilance systems of the Member States together form the pharmacovigilance system in the EU, co-operating in a network structure under the co-ordination of the EMEA and in liaison with the European Commission. Also included are Norway, Iceland and Liechtenstein, which are not members of the EU but are part of the European Economic Area (EEA) (EEA Joint Committee, 1999). Within this network structure, all parties have their roles and responsibili-ties for the surveillance of medicinal products. These roles and responsibilities vary depending on the route of marketing authorisation of the product in the EU and are defined in Directive 2001/83/EC, as amended in 2004 as a result of an intensive legislative review process, and Council Regulation (EEC) No. 2309/93, replaced, likewise through the review process, as of 20 November 2005 by Regulation (EC) No. 726/2004. They are further described in guidance documents which were developed at EU level during the 1990s for the competent authorities and marketing authorisa-tion holders in consultation with Member States and interested parties (Table 14.3). These guidelines are in accordance with recommendations agreed at the Inter-national Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). They have been amended in the light of experience and are available in a compiled format (European Commission, 2006).

The
EMEA is a Community agency, that is a public authority of the EU, set up by a
Community act of secondary legislation (Council Regulation (EEC) No. 2309/93)
with its own legal personality (European Union institutions and other bodies,
2005). The objec-tive of the EMEA is the protection and promotion of human and
animal health in the EU by fulfilling, inter
alia, the following tasks with respect to human medicines:
·
the co-ordination of the scientific evaluation of qual-ity, safety
and efficacy of medicinal products that have been applied for a central
marketing authorisa-tion with the aim of facilitating the access to effective
and safe innovative medicinal products throughout the EU; and
·
the co-ordination of post-authorisation safety of medicinal
products through the pharmacovigilance network.
·
The EMEA pools scientific expertise from the Member States
for the evaluation of medicinal prod-ucts, and to provide advice on drug
research and development programmes (European Medicines Agency, 2005). More
specific to pharmacovigi-lance, the tasks of the EMEA include the following:
–
co-ordination of the supervision (including
phar-macovigilance activities) of medicinal products authorised in the EU;
–
provision of access to information on suspected ADRs
reported for medicinal products marketed in the EU by means of a database and
data-processing network (EudraVigilance);
–
maintenance of and variations to the terms of the marketing
authorisation for centrally authorised products and
–
management of referral procedures for nationally authorised
products leading to Commission Deci-sions binding in all Member States when
there is a safety concern which impacts on public health in the Community; and
provision of recommen-dations on measures necessary to ensure safe and
effective use of these products.
EudraVigilance
was put in place by the EMEA from December 2001 (European Medicines Agency,
2004), enabling the electronic transmission of ADR case reports to a central
point accessible by all competent authorities in the EU and exchange of
pharmacovig-ilance information between all stakeholders (market-ing
authorisation holders, national competent authori-ties and EMEA). In addition
to the case reports aris-ing worldwide post-marketing, EudraVigilance was
extended to include clinical trials data as of May 2004. These developments are
in line with international devel-opments at ICH level (Tsintis and LaMache,
2004) and proactive pharmacovigilance and risk management (Waller and Evans,
2003). Guidance for the electronic submission of case reports on ADRs in
relation to medicinal products authorised in the EU is provided (European
Commission, 2006).
Much
of the work of the EMEA is done within its scientific committees. For medicines
used in humans this is the CHMP. This committee is supported by several expert
working parties, one of which is the PhVWP. The PhVWP currently meets eleven
times per year at the EMEA. Its mission is to provide advice on the safety of
medicinal products and the investigation of ADRs to enable effective risk
identification, assessment and management, in the pre- and post-authorisation
phase, leading to recommendations on harmonised and synchronised action. These
are ultimately imple-mented either by the European Commission following a CHMP
Opinion for centrally authorised products or by national competent authorities.
The PhVWP also takes the lead in the development of pharmacovigilance
guidelines.
To
facilitate, in addition, a continuous exchange of information between
regulators in the EU, in particu-lar with regard to changes in the benefit–risk
balance possibly requiring major regulatory action, but also for signal
evaluation, the so-called rapid alert-non-urgent information system has been
established. Records of this information flow are maintained centrally by the
EMEA and followed up by the PhVWP at each of their meetings. The principles and
procedures of this system are presented in a guideline (European Commission,
2006).
Pharmaceutical
companies holding marketing autho-risations in the EU have various obligations
in the area of pharmacovigilance that are laid down in Title IX of Directive
2001/83/EC and Regulation (EC) No 726/2004 and elaborated further in guidelines
(Euro-pean Commission, 2006). In particular, marketing authorisation holders
must employ a qualified person who is responsible for
·
establishing and maintaining a system that collects and
collates all suspected ADRs;
·
the preparation of periodic safety update reports;
·
responding to requests for additional information from
competent authorities; and
·
provision to competent authorities of any other infor-mation
relevant to the risk-benefit evaluation.
In
addition, marketing authorisation holders are obliged to report serious
suspected ADRs in accordance with the legislation and guidance cited above to
competent authorities within 15 days (‘expedited reports’).
Related Topics
