Mechanisms of Drug Absorption
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Absorption of Drugs
The three broad categories of drug transport mechanisms involved in absorption are – A. Transcellular/intracellular transport B. Paracellular/intercellular transport C. Vesicular transport
MECHANISMS OF DRUG ABSORPTION
The three broad categories of drug transport mechanisms involved in absorption are –
A. Transcellular/intracellular transport
B. Paracellular/intercellular transport
C. Vesicular transport
A. Transcellular/Intracellular Transport
Transcellular/Intracellular Transport – is defined as the passage of drugs across the GI epithelium. It is the most common pathway for drug transport. The 3 steps involved in transcellular transport of drugs are –
(i) Permeation of GI epithelial cell membrane, a lipoidal barrier – this is the major obstacle to drug absorption.
(ii) Movement across the intracellular space (cytosol).
(iii) Permeation of the lateral or basolateral membrane- this is of secondary importance.
The various transcellular transport processes involved in drug absorption are –
1. Passive Transport Processes – These transport processes do not require energy other than that of molecular motion (Brownian motion) to pass through the lipid bilayer. Passive transport processes can be further classified into following types –
a. Passive diffusion.
b. Pore transport.
c. Ion-pair transport.
d. Facilitated- or mediated-diffusion.
2. Active Transport Processes – This transport process requires energy from ATP to move drug molecules from extracellular to intracellular milieu. These are of two types –
a. Primary active transport.
b. Secondary active transport – this process is further subdivided into two –
i. Symport (co-transport).
ii. Antiport (counter-transport).
B. Paracellular/Intercellular Transport
Paracellular/Intercellular Transport – is defined as the transport of drugs through the junctions between the GI epithelial cells. This pathway is of minor importance in drug absorption. The two paracellular transport mechanisms involved in drug absorption are –
1. Permeation through tight junctions of epithelial cells – this process basically occurs through openings which are little bigger than the aqueous pores. Compounds such as insulin and cardiac glycosides are taken up this mechanism.
2. Persorption – is permeation of drug through temporary openings formed by shedding of two neighbouring epithelial cells into the lumen.
Paracellular transport differs from pore transport in that the former involves transfer of drug across epithelium and through the cellular junctions whereas in the case of latter, the molecules are transferred from outside of the epithelial cell into the cell through pores present in the cell membrane.
C. Vesicular or Corpuscular Transport (Endocytosis)
Vesicular or Corpuscular Transport (Endocytosis) – Like active transport, these are also energy dependent processes but involve transport of substances within vesicles into a cell. Since the mechanism involves transport across the cell membrane, the process can also be classified as transcellular. Vesicular transport of drugs can be classed into two categories –
1. Pinocytosis.
2. Phagocytosis.
Figure 2.3. compares transcellular, paracellular and vesicular transport mechanisms.
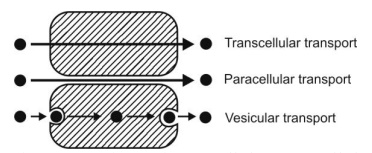
Fig. 2.3. Illustrative comparison of transcellular, paracellular and vesicular transport.
Passive Diffusion
Also called non-ionic diffusion, it is the major process for absorption of more than 90% of the drugs. The driving force for this process is the concentration or electrochemical gradient. It is defined as the difference in the drug concentration on either side of the membrane. Drug movement is a result of the kinetic energy of molecules. Since no energy source is required, the process is called as passive diffusion. During passive diffusion, the drug present in the aqueous solution at the absorption site partitions and dissolves in the lipid material of the membrane and finally leaves it by dissolving again in an aqueous medium, this time at the inside of the membrane.
Passive diffusion is best expressed by Fick’s first law of diffusion, which states that the drug molecules diffuse from a region of higher concentration to one of lower concentration until equilibrium is attained and that the rate of diffusion is directly proportional to the concentration gradient across the membrane. It can be mathematically expressed by the following equation:
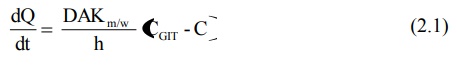
dQ/dt = rate of drug diffusion (amount/time). It also represents the rate of appearance of drug in blood
D = diffusion coefficient of the drug through the membrane (area/time)
A = surface area of the absorbing membrane for drug diffusion (area)
Km/w = partition coefficient of the drug between the lipoidal membrane and the aqueous GI fluids (no units)
(CGIT – C) = difference in the concentration of drug in the GI fluids and the plasma, called as the concentration gradient (amount/volume)
h = thickness of the membrane (length)
Based on the above equation, certain characteristics of passive diffusion can be generalized –
1. The drug moves down the concentration gradient indicating downhill transport.
2. The process is energy-independent and non-saturable.
3. The rate of drug transfer is directly proportional to the concentration gradient between GI fluids and the blood compartment.
4. Greater the area and lesser the thickness of the membrane, faster the diffusion; thus, more rapid is the rate of drug absorption from the intestine than from the stomach.
5. The process is rapid over short distances and slower over long distances.
6. Equilibrium is attained when the concentration on either side of the membrane becomes equal.
7. Drugs which can exist in both ionised and unionised forms approach equilibrium primarily by the transfer of the unionised species; the rate of transfer of unionised species is 3 to 4 times the rate for ionised drugs.
8. Greater the membrane/water partition coefficient of drug, faster the absorption; since the membrane is lipoidal in nature, a lipophilic drug diffuses at a faster rate by solubilising in the lipid layer of the membrane.
9. The drug diffuses rapidly when the volume of GI fluid is low; conversely, dilution of GI fluids decreases the drug concentration in these fluids (CGIT) and lower the concentration gradient (CGIT – C). This phenomenon is, however, made use of in treating cases of oral overdose or poisoning.
10. The process is dependent, to a lesser extent, on the square root of the molecular size of the drug – drugs having molecular weights between 100 to 400 Daltons are effectively absorbed passively. The diffusion generally decreases with increase in the molecular weight of the compound. However, there are exceptions—for example, cyclosporin A, a peptide of molecular weight 1200, is absorbed orally much better than any other peptide.
Initially, when the drug is ingested, CGIT >> C and a large concentration gradient exists thereby acting as the driving force for absorption. As equilibrium approaches, the drug diffusion should stop and consequently a large fraction of drug may remain unabsorbed. But this is not the case; once the passively absorbed drug enters blood, it is rapidly swept away and distributed into a much larger volume of body fluids and hence, the concentration of drug at the absorption site, CGIT, is maintained greater than the concentration of drug in plasma. Such a condition is called as sink condition for drug absorption.
Since under usual conditions of absorption, D, A, Km/w and h are constants, the term DAKm/w/h can be replaced by a combined constant P called as permeability coefficient. Permeability refers to the ease with which a drug can penetrate or diffuse through a membrane. Moreover, due to sink conditions, the concentration of drug in plasma C is very small in comparison to CGIT. As a result, equation 2.1. may be simplified to:
dQ / dt = PCGIT

Equation 2.2 is an expression for a first-order process. Thus, passive diffusion follows first-order kinetics. Since a large concentration gradient always exists at the absorption site for passive diffusion, the rate of drug absorption is usually more rapid than the rate of elimination. Besides, dilution and distribution of the absorbed drug into a large pool of body fluids and its subsequent binding to various tissues are other reasons for elimination being slower than absorption.
Figure 2.4 illustrates the relative permeability of different molecules to lipid bilayer.
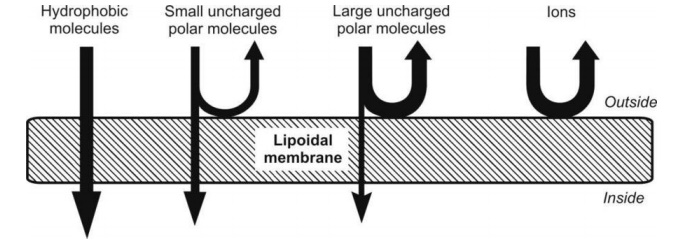
Fig. 2.4. Relative passive diffusion rate of different types of molecules
Pore Transport
It is also called as convective transport, bulk flow or filtration. This mechanism is responsible for transport of molecules into the cell through the protein channels present in the cell membrane. Following are the characteristics of pore transport –
1. The driving force is constituted by the hydrostatic pressure or the osmotic differences across the membrane due to which bulk flow of water along with small solid molecules occurs through such aqueous channels. Water flux that promotes such a transport is called as solvent drag.
2. The process is important in the absorption of low molecular weight (less than 100), low molecular size (smaller than the diameter of the pore) and generally water-soluble drugs through narrow, aqueous-filled channels or pores in the membrane structure—for example, urea, water and sugars.
3. Chain-like or linear compounds of molecular weight up to 400 Daltons can be absorbed by filtration.
Drug permeation through water-filled channels is of particular importance in renal excretion, removal of drug from the cerebrospinal fluid and entry of drugs into the liver.
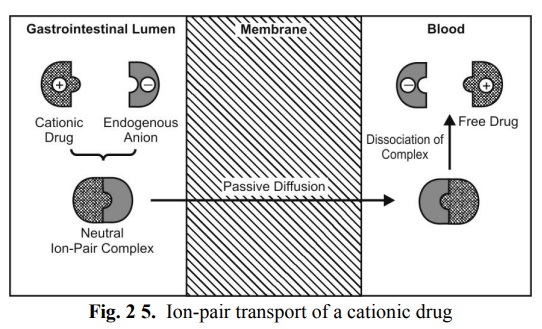
Ion-Pair Transport
Yet another mechanism that explains the absorption of drugs like quaternary ammonium compounds and sulphonic acids, which ionise under all pH conditions, is ion-pair transport. Despite their low o/w partition coefficient values, such agents penetrate the membrane by forming reversible neutral complexes with endogenous ions of the GIT like mucin. Such neutral complexes have both the required lipophilicity as well as aqueous solubility for passive diffusion. Such a phenomenon is called as ion-pair transport (Fig. 2.5). Propranolol, a basic drug that forms an ion pair with oleic acid, is absorbed by this mechanism.
Carrier-Mediated Transport
Some polar drugs cross the membrane more readily than can be predicted from their concentration gradient and partition coefficient values. This suggests presence of specialized transport mechanisms without which many essential water-soluble nutrients like monosaccharides, amino acids and vitamins will be poorly absorbed. The mechanism is thought to involve a component of the membrane called as the carrier that binds reversibly or non-covalently with the solute molecules to be transported. This carrier-solute complex traverses across the membrane to the other side where it dissociates and discharges the solute molecule. The carrier then returns to its original site to complete the cycle by accepting a fresh molecule of solute. Carriers in membranes are proteins (transport proteins) and may be an enzyme or some other component of the membrane. They are numerous in all biological membranes and are found dissolved in the lipid bilayer of the membrane.
Important characteristics of carrier-mediated transport are:
1. A carrier protein always has an uncharged (non-polar) outer surface which allows it to be soluble within the lipid of the membrane.
2. The carriers have no directionality; they work with same efficiency in both directions.
3. The transport process is structure-specific i.e. the carriers have special affinity for and transfer a drug of specific chemical structure only (i.e. lock and key arrangement); generally the carriers have special affinity for essential nutrients.
4. Since the system is structure-specific, drugs having structure similar to essential nutrients, called as false nutrients, are absorbed by the same carrier system. This mechanism is of particular importance in the absorption of several antineoplastic agents like 5-fluorouracil and 5-bromouracil which serve as false nutrients.
5. As the number of carriers is limited, the transport system is subject to competition between agents having similar structure.
6. Since the number of carriers is limited, the system is capacity-limited i.e. at higher drug concentration; the system becomes saturated and approaches an asymptote. It is important to note that for a drug absorbed by passive diffusion, the rate of absorption increases linearly with the concentration but in case of carrier-mediated processes, the drug absorption increases linearly with concentration until the carriers become saturated after which it becomes curvilinear and approach a constant value at higher doses (see Fig. 2.6). Such a capacity-limited process can be adequately described by mixed order kinetics, also called as Michaelis-Menten, saturation or non-linear kinetics. The process is called mixed-order because it is first-order at sub-saturation drug concentrations and apparent zero-order at and above saturation levels. Moreover, the capacity-limited characteristics of such a system suggest that the bioavailability of a drug absorbed by such a system decrease with increasing dose—for example, vitamins like B1, B2 and B12. Hence, administration of a large single oral dose of such vitamins is irrational.
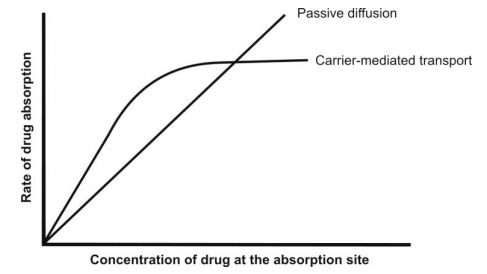
Fig. 2.6. Comparison of rate of absorption versus drug concentration plots for passive and carrier-mediated transport processes
5. Specialized absorption or carrier-mediated absorption generally occurs from specific sites of the intestinal tract which are rich in number of carriers. Such an area in which the carrier system is most dense is called as absorption window. Drugs absorbed through such absorption windows are poor candidates for controlled release formulations.
Two types of carrier-mediated transport systems have been identified.
They are—facilitated diffusion and active transport.
Facilitated Diffusion
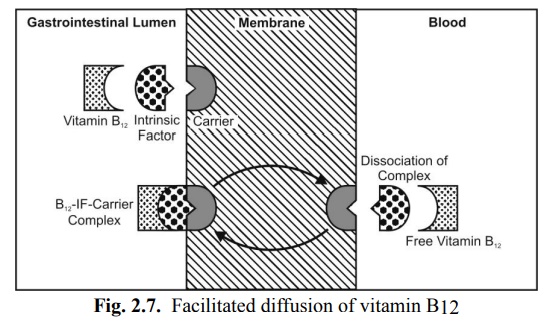
It is a carrier-mediated transport system that operates down the concentration gradient (downhill transport) but at a much a faster rate than can be accounted by simple passive diffusion. The driving force is concentration gradient (hence a passive process). Since no energy expenditure is involved, the process is not inhibited by metabolic poisons that interfere with energy production. Facilitated diffusion is of limited importance in the absorption of drugs. Examples of such a transport system include entry of glucose into RBCs and intestinal absorption of vitamins B1 and B2. A classic example of passive facilitated diffusion is the GI absorption of vitamin B12. An intrinsic factor (IF), a glycoprotein produced by the gastric parietal cells, forms a complex with vitamin B12 which is then transported across the intestinal membrane by a carrier system (Fig. 2.7).
Active Transport
This transport mechanism requires energy in the form ATP. Active transport mechanisms are further subdivided into -
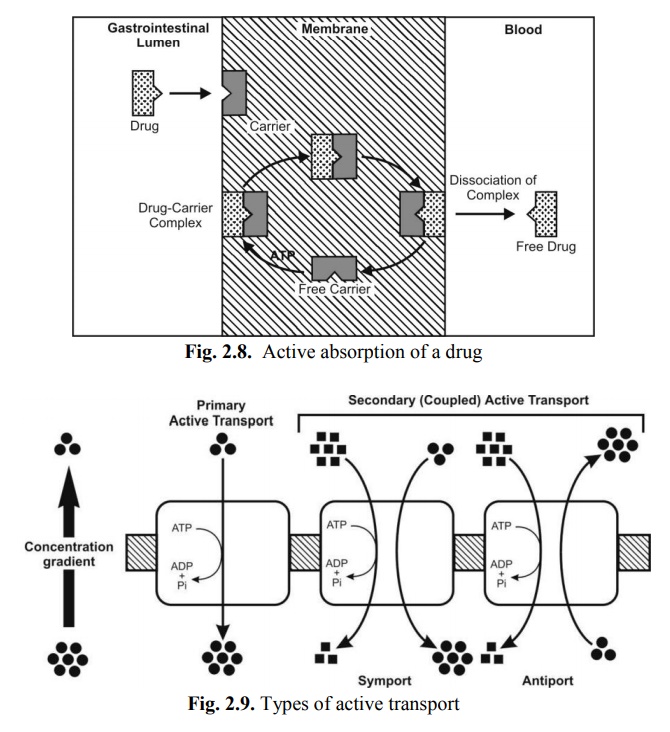
a. Primary active transport – In this process, there is direct ATP requirement. Moreover, the process transfers only one ion or molecule and in only one direction, and hence called as uniporter e.g. absorption of glucose. Carrier proteins involved in primary active transport are of two types –
(i) Ion transporters – are responsible for transporting ions in or out of cells. A classic example of ATP-driven ion pump is proton pump which is implicated in acidification of intracellular compartments. Two types of ion transporters which play important role in the intestinal absorption of drugs have been identified –
(a) Organic anion transporter – which aids absorption of drugs such as pravastatin and atorvastatin.
(b) Organic cation transporter – which aids absorption of drugs such as diphenhydramine.
(ii) ABC (ATP-binding cassette) transporters – are responsible for transporting small foreign molecules (like drugs and toxins) especially out of cells (and thus called as efflux pumps) which make them clinically important. A classic example of ABC transporter is P-glycoprotein (P-gp). The latter is responsible for pumping hydrophobic drugs especially anticancer drugs out of cells. Presence of large quantity of this protein thus makes the cells resistant to a variety of drugs used in cancer chemotherapy, a phenomenon called as multi-drug resistance. It is for this reason that P-gp is called as multi-drug resistance (MDR) protein. ABC transporters present in brain capillaries pump a wide range of drugs out of brain.
b. Secondary active transport – In these processes, there is no direct requirement of ATP i.e. it takes advantage of previously existing concentration gradient. The energy required in transporting an ion aids transport of another ion or molecule (co-transport or coupled transport) either in the same direction or in the opposite direction. Accordingly this process is further subdivided into –
i. Symport (co-transport) – involves movement of both molecules in the same direction e.g. Na+-glucose symporter uses the potential energy of the Na+ concentration gradient to move glucose against its concentration gradient. A classic example of symporter is peptide transporter called as H+-coupled peptide transporter (PEPT1) which is implicated in the intestinal absorption of peptide-like drugs such as β-lactam antibiotics.
ii. Antiport (counter-transport) – involves movement of molecules in the opposite direction e.g. expulsion of H+ ions using the Na+ gradient in the kidneys.
Figure 2.8 illustrates active transport of a drug and figure 2.9 represents the types of active transport.
Active transport is a more important process than facilitated diffusion in the absorption of nutrients and drugs and differs from it in several respects:
1. The drug is transported from a region of lower to one of higher concentration i.e. against the concentration gradient (in the case of ions, against an electrochemical gradient) or uphill transport, without any regard for equilibrium.
2. The process is faster than passive diffusion.
3. Since the process is uphill, energy is required in the work done by the carrier.
4. As the process requires expenditure of energy, it can be inhibited by metabolic poisons that interfere with energy production like fluorides, cyanide and dinitrophenol and lack of oxygen, etc. Endogenous substances that are transported actively include sodium, potassium, calcium, iron, glucose, certain amino acids and vitamins like niacin, pyridoxin and ascorbic acid. Drugs having structural similarity to such agents are absorbed actively, particularly the agents useful in cancer chemotherapy. Examples include absorption of 5-fluorouracil and 5-bromouracil via the pyrimidine transport system, absorption of methyldopa and levodopa via an L-amino acid transport system and absorption of ACE inhibitor enalapril via the small peptide carrier system. A good example of competitive inhibition of drug absorption via active transport is the impaired absorption of levodopa when ingested with meals rich in proteins. Active transport is also important in renal and biliary excretion of many drugs and their metabolites and secretion of certain acids out of the CNS.
Figure 2.10 compares active and passive transport
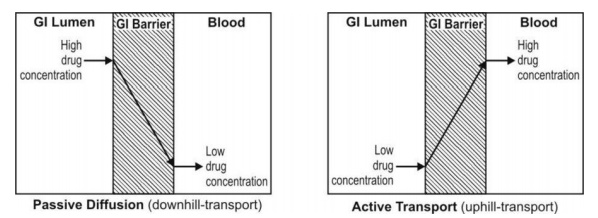
Fig. 2.10. Comparison between active and passive transport
Endocytosis
It is a minor transport mechanism which involves engulfing extracellular materials within a segment of the cell membrane to form a saccule or a vesicle (hence also called as corpuscular or vesicular transport) which is then pinched-off intracellularly (Fig. 2.11). This is the only transport mechanism whereby a drug or compound does not have to be in an aqueous solution in order to be absorbed.
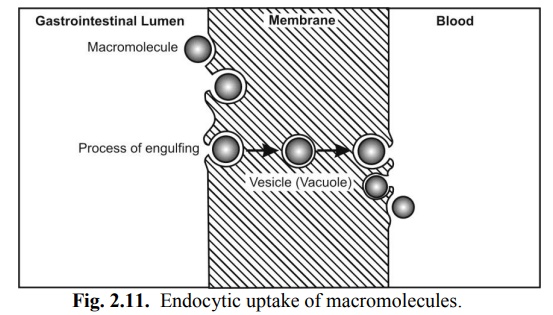
This phenomenon is responsible for the cellular uptake of macromolecular nutrients like fats and starch, oil soluble vitamins like A, D, E and K, water soluble vitamin like B12 and drugs such as insulin. Another significance of such a process is that the drug is absorbed into the lymphatic circulation thereby bypassing first-pass hepatic metabolism.
Endocytosis includes two types of processes:
1. Phagocytosis (cell eating): adsorptive uptake of solid particulates, and
2. Pinocytosis (cell drinking): uptake of fluid solute.
Orally administered Sabin polio vaccine, large protein molecules and the botulism toxin (that causes food poisoning) are thought to be absorbed by pinocytosis. Sometimes, an endocytic vesicle is transferred from one extracellular compartment to another. Such a phenomenon is called as transcytosis.
Combined Absorption Mechanisms
A drug might be absorbed by more than just one mechanism—for example, cardiac glycosides are absorbed both passively as well as by active transport. Vitamin B12 is absorbed by passive diffusion, facilitated diffusion as well as endocytosis. The transport mechanism also depends upon the site of drug administration (see Table 2.8).
Absorption of drugs by various mechanisms is summarized in Fig. 2.12.
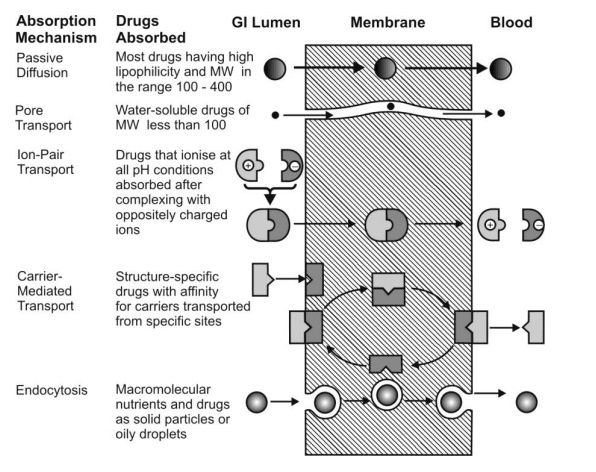
Fig. 2.12. Summary of important transport processes and drugs absorbed through them
Phases of Drug Transfer from GI Absorption Site (GI Epithelium) into Systemic Circulation
Absorption of drugs through the GI epithelium can be divided into three phases –
1. Pre-uptake phase – the two important pre-uptake processes are –
(a) Dissolution of drug in the GI fluids.
(b) Metabolism of drug in the GI lumen – this can be affected by –
(i) Digestive enzymes present in the GIT, and/or
(ii) Bacterial enzymes in the colon.
2. Uptake phase – is three processes involved in drug uptake are –
(a) Delivery of drug to the absorption site in the GIT.
(b) Metabolism of drug by enzymes in the GI epithelium (gut wall metabolism).
(c) Passage of drug through the GI epithelium.
3. Post-uptake phase - the three important post-uptake processes are –
(a) Metabolism of drug by the liver, en route to the systemic circulation (first-pass hepatic metabolism).
(b) Enterohepatic circulation of drug – during the first pass through the liver, the drug may be excreted in the bile, re-enter the GIT via gall bladder and gets reabsorbed.
(c) Transfer of drug into the systemic circulation.
Routes of Drug Transfer from the Absorption Site in GIT into the Systemic Circulation
A drug is transferred from the absorption site into systemic circulation by one of the two routes –
1. Splanchnic circulation – which is the network of blood vessels that supply the GIT. It is the major route for absorption of drug into the systemic circulation. A drug that enters splanchnic circulation goes to the liver first where it may undergo presystemic metabolism before finally arriving into the systemic circulation. A drug whose uptake is through stomach, small intestine or large intestine goes into the systemic circulation via splanchnic circulation. Rectally administered drugs have direct access to systemic circulation and thus circumvent firs-pass effect.
2. Lymphatic circulation – is a path of minor importance in drug absorption into systemic circulation for two reasons –
(a) The lymph vessels are less accessible than the capillaries
(b) The lymph flow is exceptionally slow.
However, fats, fat-soluble vitamins and highly lipophilic drugs are absorbed through lymphatic circulation.
There are three advantages of lymphatic absorption of drugs –
(a) Avoidance of first-pass effect.
(b) Compounds of high molecular weight (above 16,000) can be absorbed by lymphatic transport.
(c) Targeted delivery of drugs to lymphatic system as in certain cases of cancer is possible.
Figure 2.13 represents the transfer of drug to splanchnic and lymphatic circulation after its uptake by the intestinal epithelium.
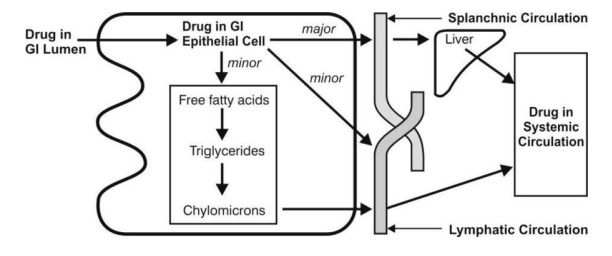
Fig. 2.13. Transfer of drug from intestinal epithelium to splanchnic and lymphatic circulation
Related Topics
