Patient Related Factors Affecting Drug Absorption
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Absorption of Drugs
include factors relating to the anatomical, physiological and pathological characteristics of the patient
PATIENT RELATED FACTORS
include factors relating to the anatomical, physiological and pathological characteristics of the patient
1. Age
2. Gastric emptying time
3. Intestinal transit time
4. Gastrointestinal pH
5. Disease states
6. Blood flow through the GIT
7. Gastrointestinal contents:
a. Other drugs
b. Food
c. Fluids
d. Other normal GI contents
8. Presystemic metabolism by:
a. Luminal enzymes
b. Gut wall enzymes
c. Bacterial enzymes
d. Hepatic enzymes
1. Age
In infants, the gastric pH is high and intestinal surface and blood flow to the GIT is low resulting in altered absorption pattern in comparison to adults. In elderly persons, causes of impaired drug absorption include altered gastric emptying, decreased intestinal surface area and GI blood flow, higher incidents of achlorhydria and bacterial overgrowth in small intestine.
2. Gastric Emptying
Apart from dissolution of a drug and its permeation through the biomembrane, the passage from stomach to the small intestine, called as gastric emptying, can also be a rate-limiting step in drug absorption because the major site of drug absorption is intestine. Thus, generally speaking, rapid gastric emptying increases bioavailability of a drug.
Rapid gastric emptying is advisable where:
1. A rapid onset of action is desired e.g. sedatives.
2. Dissolution of drug occurs in the intestine e.g. enteric-coated dosage forms.
3. The drugs are not stable in the gastric fluids e.g. penicillin G and erythromycin.
4. The drug is best absorbed from the distal part of the small intestine e.g. vitamin B12.
For better dissolution and absorption, the gastric emptying can be promoted by taking the drug on empty stomach. Since gastric emptying is altered by several factors due to which large intersubject variations are observed, all biopharmaceutic studies that require the drug to be taken orally are performed in volunteers on empty stomach.
Gastric emptying of a drug is delayed by co-administering food because unless the gastric contents are fluid enough or the size of the solid particles is reduced below 2 mm, its passage through the pylorus into the intestine is not possible.
Delay in gastric emptying is recommended in particular where:
1. The food promotes drug dissolution and absorption e.g. griseofulvin.
2. Disintegration and dissolution of dosage form is promoted by gastric fluids.
3. The drugs dissolve slowly e.g. griseofulvin.
4. The drugs irritate the gastric mucosa e.g. aspirin, phenylbutazone and nitrofurantoin.
5. The drugs are absorbed from the proximal part of the small intestine and prolonged drug-absorption site contact is desired e.g. vitamin B2 and vitamin C
Gastric emptying is a first-order process. Several parameters are used to quantify gastric emptying:
1. Gastric emptying rate is the speed at which the stomach contents empty into the intestine.
2. Gastric emptying time is the time required for the gastric contents to empty into the small intestine. Longer the gastric emptying time, lesser the gastric emptying rate.
3. Gastric emptying t½ is the time taken for half the stomach contents to empty.
In vivo gastric emptying of a drug (so also the disintegration of dosage form and drug release) can be studied by using radio-opaque contrast materials like barium sulphate or tagging the drug with a radioisotope and scanning the stomach at regular intervals of time.
A large number of factors influence gastric emptying as discussed below.
1. Volume of meal: Larger the bulk of the meals, longer the gastric emptying time. However, an initial rapid rate of emptying is observed with a large meal volume and an initial lag phase in emptying of a small volume meal. Since gastric emptying is a first-order process, a plot of log of volume of contents remaining in the stomach versus time yields a straight line.
2. Composition of meal: Predictably, the rate of gastric emptying for various food materials is in the following order: carbohydrates > proteins > fats. Fats promote secretion of bile which too has an inhibitory effect on gastric emptying. Delayed gastric emptying as observed with fatty meals, is beneficial for the absorption of poorly soluble drugs like griseofulvin.
3. Physical state and viscosity of meal: Liquid meals take less than an hour to empty whereas a solid meal may take as long as 6 to 7 hours. Viscous materials empty at a slow rate in comparison to less viscous materials.
4. Temperature of the meal: High or low temperature of the ingested fluid (in comparison to body temperature) reduce the gastric emptying rate.
5. Gastrointestinal pH: Gastric emptying is retarded at low stomach pH and promoted at higher or alkaline pH. Chemicals that affect gastrointestinal pH also alter gastric emptying. The inhibitory effect of various acids on gastric emptying decreases with increase in molecular weight and is in the following order -
HCl > acetic > lactic > tartaric > citric
With alkaline solutions, a low base concentration (1% NaHCO3) increases the gastric emptying rate more than the one of higher concentration (5%).
6. Electrolytes and osmotic pressure: Water, isotonic solutions, and solutions of low salt concentration empty the stomach rapidly whereas a higher electrolyte concentration decreases gastric emptying rate.
7. Body posture: Gastric emptying is favoured while standing and by lying on the right side since the normal curvature of the stomach provides a downhill path whereas lying on the left side or in supine position retards it.
8. Emotional state: Stress and anxiety promote gastric motility whereas depression retards it.
9. Exercise: Vigorous physical training retards gastric emptying.
10. Disease states: Diseases like gastroenteritis, gastric ulcer, pyloric stenosis, diabetes and hypothyroidism retard gastric emptying. Partial or total gastrectomy, duodenal ulcer and hyperthyroidism promote gastric emptying rate.
11. Drugs: Drugs that retard gastric emptying include poorly soluble antacids (aluminium hydroxide), anticholinergics (atropine, propantheline), narcotic analgesics (morphine) and tricyclic antidepressants (imipramine, amitriptyline). Metoclopramide, dom-peridone and cisapride (prokinetic agents) stimulate gastric emptying.
The passage of drug through the oesophagus, called as oesophageal transit, is important in persons who swallow the solid dosage form lying down in supine position or with little or no water. In such cases, the dosage form remains lodged in the oesophagus and disintegrates slowly which may result in delayed absorption or local damage to the mucosa from drugs like NSAIDs.
3. Intestinal Transit
Since small intestine is the major site for absorption of most drugs, long intestinal transit time is desirable for complete drug absorption (see Table 2.8).
TABLE 2.8.
Transit Time for Contents from Different Regions of Intestine
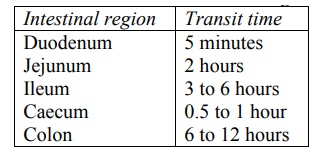
The residence time depends upon the intestinal motility or contractions. The mixing movement of the intestine that occurs due to peristaltic contractions promote drug absorption, firstly, by increasing the drug-intestinal membrane contact, and secondly, by enhancing drug dissolution especially of poorly soluble drugs, through induced agitation.
Delayed intestinal transit is desirable for:
1. Drugs that dissolve or release slowly from their dosage form (sustained-release products) or when the ratio of dose to solubility is high e.g. chlorothiazide.
2. Drugs that dissolve only in the intestine (enteric-coated formulations).
3. Drugs which are absorbed from specific sites in the intestine (several B vitamins, lithium carbonate, etc.).
4. When the drug penetrates the intestinal mucosa very slowly e.g. acyclovir.
5. When absorption of drug from the colon is minimal.
However, as the contents move down the intestine into the colon, its viscosity gradually increases due to absorption of water and electrolytes which limits the design of sustained release products of drugs having short biological half-lives.
Like gastric emptying, intestinal transit is influenced by several factors like food, drugs and diseases. Food, decreased digestive secretions and pregnancy retard intestinal transit whereas diarrhoea promotes it. Drugs like metoclopramide that promote gastric emptying and intestinal transit enhance absorption of rapidly soluble drugs. Laxatives also promote the rate of intestinal transit. Drugs such as anticholinergics that retard gastric and intestinal transit promote absorption of poorly soluble drugs—for example, propantheline shows a 100%, 50% and 30% rise in the absorption of vitamin B2, nitrofurantoin and hydrochlorothiazide respectively.
4. Gastrointestinal pH
A tremendous 107 fold difference in the hydrogen ion concentration is observed between the gastric and colon fluids. The GI pH generally increases gradually as one move down the stomach to the colon and rectum (see Fig. 2.22). GI fluid pH influence drug absorption in several ways:
1. Disintegration: The disintegration of some dosage forms is pH sensitive. With enteric-coated formulations, the coat dissolves only in the intestine followed by disintegration of the tablet.
2. Dissolution: A large number of drugs are either weak acids or weak bases whose solubility is greatly affected by pH. A pH that favours formation of salt of the drug enhances the dissolution of that drug. Since drug dissolution is one of the important rate-determining steps in drug absorption, GI pH is of great significance in the oral bioavailability of drugs. Weakly acidic drugs dissolve rapidly in the alkaline pH of the intestine whereas basic drugs dissolve in the acidic pH of the stomach. Since the primary site for absorption of most drugs is small intestine, the poorly water-soluble basic drugs must first dissolve in the acidic pH of stomach before moving into the intestine.
3. Absorption: Depending upon the drug pKa and whether its an acidic or a basic drug, the GI pH influences drug absorption by determining the amount of drug that would exist in the unionised form at the site of absorption. This topic has already been dealt with in sufficient details under pH-partition hypothesis.
4. Stability: GI pH also influences the chemical stability of drugs. The acidic stomach pH is known to affect degradation of penicillin G and erythromycin. This can be overcome by preparing prodrugs of such drugs that do not degrade or dissolve in acidic pH e.g. carindacillin and erythromycin estolate. With basic drugs, formation of insoluble drug hydroxide in the alkaline pH of the intestine has been observed.
5. Disease States
Several disease states can influence the rate and extent of drug absorption. The 3 major classes of disease states that can influence the bioavailability of a drug are:
1. Gastrointestinal diseases,
2. Cardiovascular diseases, and
3. Hepatic diseases.
1. Gastrointestinal diseases: A number of pathologic conditions of the GIT can influence changes in drug absorption pattern, namely:
(a) Altered GI motility: (discussed earlier)
(b) Gastrointestinal diseases and infections: The influence of achlorhydria (decreased gastric acid secretion and increased stomach pH) on gastric emptying and drug absorption, especially that of acidic drugs (decreased absorption, e.g. aspirin) has been studied. Two of the intestinal disorders related with malabsorption syndrome that influence drug availability are celiac disease (characterized by destruction of villi and microvilli) and Crohn’s disease. Abnormalities associated with celiac disease include increased gastric emptying rate and GI permeability, altered intestinal drug metabolism, steatorrhea (impaired secretion of bile thus affecting absorption of lipophilic drugs and vitamins) and reduced enterohepatic cycling of bile salts, all of which can significantly impair drug absorption. Conditions associated with Crohn’s disease that can alter absorption pattern are altered gut wall microbial flora, decreased gut surface area and intestinal transit rate. Malabsorption is also induced by drugs such as antineoplastics and alcohol which increase permeability of agents not normally absorbed. GI infections like shigellosis, gastroenteritis, cholera and food poisoning also result in malabsorption. Colonic diseases such as colitis, amoebiasis and constipation can also alter drug absorption.
(c) Gastrointestinal surgery: Gastrectomy can result in drug dumping in the intestine, osmotic diarrhoea and reduced intestinal transit time. Intestinal surgery also influences drug absorption for predictable reasons.
2. Cardiovascular diseases: Several changes associated with congestive cardiac failure influence bioavailability of a drug viz. oedema of the intestine, decreased blood flow to the GIT and gastric emptying rate and altered GI pH, secretions and microbial flora.
3. Hepatic diseases: Disorders such as hepatic cirrhosis influence bioavailability mainly of drugs that undergo considerable first-pass hepatic metabolism e.g. propranolol; enhanced bioavailability is observed in such cases.
6. Blood Flow to the GIT
The GIT is extensively supplied by blood capillary network and the lymphatic system. The absorbed drug can thus be taken up by the blood or the lymph. Since the blood flow rate to the GIT (splanchnic circulation) is 500 to 1000 times (28% of cardiac output) more than the lymph flow, most drugs reach the systemic circulation via blood whereas only a few drugs, especially low molecular weight, lipid soluble compounds are removed by lymphatic system.
The high perfusion rate of GIT ensures that once the drug has crossed the membrane, it is rapidly removed from the absorption site thus maintaining the sink conditions and concentration gradient for continued drug absorption.
For drugs that have high permeation rates, e.g. highly lipid soluble drugs or drugs absorbed through pores, the GI perfusion rate could be a rate-limiting step in the absorption, e.g. tritiated water. This is not so in the case of drugs having poor permeability coefficient, e.g. ribitol. Blood flow is also important for actively absorbed drugs since oxygen and energy is required for transportation.
Food influences blood flow to the GIT. The perfusion rate increases after meals and persists for few hours but drug absorption is not influenced significantly.
7. Gastrointestinal Contents
A number of GI contents can influence drug absorption as discussed below:
1. Food-drug interactions: Presence of food may either delay, reduce, increase or may not affect drug absorption (Table 2.9).
TABLE 2.9.
Influence of Food on Drug Absorption
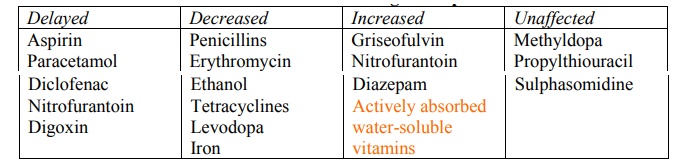
Food-drug interactions may be due to the influence of food on physiologic functions (alterations in the GI emptying rate, GI fluid secretions, pH, blood flow and absorptive processes) and/or a consequence of physicochemical interaction with the drug (alteration in drug dissolution profile, complexation and adsorption).
As a general rule, drugs are better absorbed under fasting conditions and presence of food retards or prevents it. Food does not significantly influence absorption of a drug taken half an hour or more before meals and two hours or more after meals.
Delayed or decreased drug absorption by the food could be due to one or more of the several mechanisms:
(a) Delayed gastric emptying, affecting drugs unstable in the stomach e.g. penicillins, or preventing the transit of enteric coated tablets into the intestine which may be as long as 6 to 8 hours
(b) Formation of a poorly soluble, unabsorbable complex e.g. tetracycline-calcium
(c) Increased viscosity due to food thereby preventing drug dissolution and/or diffusion towards the absorption site
Increased drug absorption following a meal could be due to one or more of the under mentioned reasons –
(a) Increased time for dissolution of a poorly soluble drug
(b) Enhanced solubility due to GI secretions like bile
(c) Prolonged residence time and absorption site contact of the drug e.g. water-soluble vitamins
(d) Increased lymphatic absorption e.g. acitretin
The specific meal components also have an influence on drug absorption. Meals high in fat aid solubilisation of poorly aqueous soluble drugs e.g. isotretinoin. Food high in proteins inhibits absorption of levodopa. An interesting example of influence of high protein meal is that of increased oral availability of propranolol to which two reasons were attributed—firstly, such a meal increases the hepatic blood flow due to which the drug can bypass first-pass hepatic metabolism (propranolol is a drug with high hepatic extraction), and secondly, it promotes blood flow to the GIT thus aiding drug absorption.
2. Fluid volume: Administration of a drug with large fluid volume results in better dissolution, rapid gastric emptying and enhanced absorption—for example, erythromycin is better absorbed when taken with a glass of water under fasting condition than when taken with meals.
3. Interaction of drug with normal GI constituents: The GIT contains a number of normal constituents such as mucin, bile salts and enzymes which influence drug absorption. Mucin, a protective mucopolysaccharide that lines the GI mucosa, interacts with streptomycin and certain quaternary ammonium compounds and retards their absorption. It also acts as a barrier to diffusion of drugs. The bile salts aid solubilisation and absorption of lipid soluble drugs like griseofulvin and vitamins A, D, E and K on one hand and on the other, decreases absorption of neomycin and kanamycin by forming water insoluble complexes.
The influence of GI enzymes on absorption will be discussed later.
4. Drug-Drug interactions in the GIT: Like food-drug interactions, drug-drug interactions can either be physicochemical or physiological.
(a) Physicochemical drug-drug interactions can be due to—
Adsorption: Antidiarrhoeal preparations containing adsorbents like attapulgite or kaolin-pectin retard/prevent absorption of a number of drugs co-administered with them e.g. promazine and lincomycin.
Complexation: Antacids and/or mineral substitutes containing heavy metals such as aluminium, calcium, iron, magnesium or zinc retard absorption of tetracyclines through formation of unabsorbable complexes. The anion exchange resins, cholestyramine and colestipol, bind cholesterol metabolites, bile salts and a number of drugs in the intestine and prevent their absorption.
pH change: Basic drugs dissolve in gastric pH; co-administration of such drugs, e.g. tetracycline with antacids such as sodium bicarbonate results in elevation of stomach pH and hence decreases dissolution rate or causes precipitation of drug.
(b) Physiologic drug-drug interactions can be due to following mechanisms—
Decreased GI transit: Anticholinergics such as propantheline retard GI motility and promote absorption of drugs like ranitidine and digoxin, whereas delay absorption of paracetamol and sulpha-methoxazole.
Increased gastric emptying: Metoclopramide promotes GI motility and enhances absorption of tetracycline, pivampicillin and levodopa.
Altered GI metabolism: Antibiotics inhibit bacterial metabolism of drugs, e.g. erythromycin enhances efficacy of digoxin by this mechanism. Co-administration of antibiotics with oral contraceptives like ethinyl oestradiol decreases the efficacy of latter by decreasing enterohepatic cycling of steroid conjugates which otherwise are hydrolysed by gut bacteria after biliary excretion.
8. Presystemic Metabolism/First-Pass Effects
For a drug administered orally, the 3 main reasons for its decreased bioavailability are:
1. Decreased absorption (owing to adsorption, precipitation, complexation and poor solubility).
2. Destabilisation or destruction of drug.
3. First-pass/presystemic metabolism (see figure 2.30)
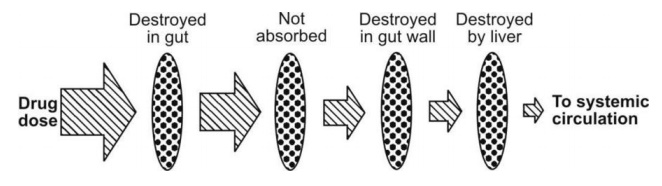
Fig. 2.30. Processes that reduce the availability of orally administered drugs
Before a drug reaches blood circulation, it has to pass for the first time through organs of elimination namely the GIT and the liver. The loss of drug through biotransformation by such eliminating organs during its passage to systemic circulation is called as first-pass or presystemic metabolism. The diminished drug concentration or rarely, complete absence of the drug in plasma after oral administration is indicative of first-pass effects. The 3 primary systems which affect presystemic metabolism of a drug are (Fig. 2.31):
1. Luminal enzymes – the metabolism by these enzymes are further categorised into two –
(a) Digestive enzymes, and
(b) Bacterial enzymes.
2. Gut wall enzymes/mucosal enzymes.
3. Hepatic enzymes.
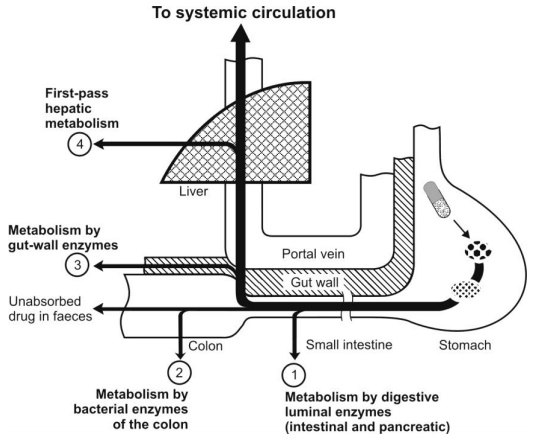
Fig. 2.31. Different sites of presystemic metabolism
1. Digestive enzymes: These are the enzymes present in the gut fluids and include enzymes from intestinal and pancreatic secretions. The latter contains hydrolases which hydrolyse ester drugs like chloramphenicol palmitate into active chloramphenicol, and peptidases which split amide linkages and inactivate protein/polypeptide drugs. Thus, one of the approaches to effect oral absorption of peptides is to deliver them to colon which lack peptidases.
2. Bacterial enzymes: The GI microflora is scantily present in stomach and small intestine and is rich in colon. Hence, most orally administered drugs remain unaffected by them. The colonic microbes generally render a drug more active or toxic on biotransformation—for example sulphasalazine, a drug used in ulcerative colitis, is hydrolysed to sulphapyridine and 5-amino salicylic acid by the microbial enzymes of the colon. An important role of intestinal microflora is that in enterohepatic cycling. Their enzymes hydrolyse the conjugates of drugs actively secreted via bile such as glucuronides of digoxin and oral contraceptives. The free drugs are reabsorbed into the systemic circulation.
3. Gut wall enzymes: Also called as mucosal enzymes, they are present in stomach, intestine and colon. Alcohol dehydrogenase (ADH) is an enzyme of stomach mucosa that inactivates ethanol. Intestinal mucosa contains both phase I and phase II (predominant) enzymes, e.g. sulphation of ethinyl oestradiol and isoprenaline. The colonic mucosa also contain both phase I and phase II enzymes. However, it is only the enzymes of the proximal small intestine that are most active.
4. Hepatic enzymes: Several drugs undergo first-pass hepatic metabolism, the highly extracted ones being isoprenaline, propranolol, alprenolol, pentoxifylline, nitroglycerine, diltiazem, nifedipine, lidocaine, morphine, etc.
Related Topics
