Physicochemical Factors Affecting Drug Absorption
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Absorption of Drugs
Physicochemical factors: 1) Drug solubility & dissolution rate 2) Particle Size and Effective Surface Area of the Drug 3) Polymorphism and Amorphism 4) Hydrates/Solvates (Pseudopolymorphism) 5) Salt form of the drug 6) Drug pKa and Lipophilicity and GI pH—pH Partition Hypothesis 7) pKa of drug & gastrointestinal pH hypothesis 8) Drug stability
PHYSICOCHEMICAL FACTORS AFFECTING DRUG ABSORPTION
1) Drug solubility & dissolution rate
2) Particle Size and Effective Surface Area of the Drug
3) Polymorphism and Amorphism
4) Hydrates/Solvates (Pseudopolymorphism)
5) Salt form of the drug
6) Drug pKa and Lipophilicity and GI pH—pH Partition Hypothesis
7) pKa of drug & gastrointestinal pH hypothesis
8) Drug stability
1. Drug Solubility and Dissolution Rate
Consider the events that occur following oral administration of a solid dosage form as shown in Fig. 2.14. Except in case of controlled-release formulations, disintegration and deaggregation occur rapidly if it is a well-formulated dosage form. Thus, the two critical slower rate-determining processes in the absorption of orally administered drugs are:
1. Rate of dissolution, and
2. Rate of drug permeation through the biomembrane.
Dissolution is the RDS for hydrophobic, poorly aqueous soluble drugs like griseofulvin and spironolactone; absorption of such drugs is often said to be dissolution rate-limited. If the drug is hydrophilic with high aqueous solubility—for example, cromolyn sodium or neomycin, then dissolution is rapid and RDS in the absorption of such drugs is rate of permeation through the biomembrane. In other words, absorption of such drugs is said to be permeation rate-limited or transmembrane rate-limited (Fig. 2.15).
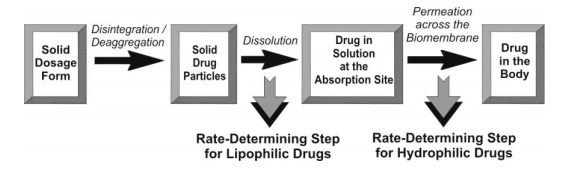
Fig. 2.15. The two rate-determining steps in the absorption of drugs from orally administered formulations
Based on the intestinal permeability and solubility of drugs, Amidon et al developed Biopharmaceutics Classification System (BCS) which classifies the drugs into one of the 4 groups as shown in the table 2.3.
TABLE 2.3.
The Biopharmaceutics Classification System for Drugs
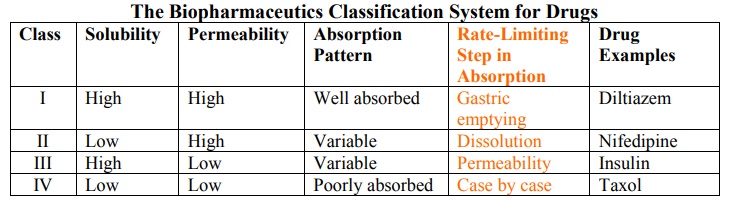
Class I drugs (high solubility/high permeability) are well absorbed orally since they have neither solubility nor permeability limitation.
Class II drugs (low solubility/high permeability) show variable absorption owing to solubility limitation.
Class III drugs (high solubility/low permeability) also show variable absorption owing to permeability limitation.
Class IV drugs (low solubility/low permeability) are poorly absorbed orally owing to both solubility and permeability limitations.
An important prerequisite for the absorption of a drug by all mechanisms except endocytosis is that it must be present in aqueous solution. This in turn depends on the drug’s aqueous solubility and its dissolution rate. Absolute or intrinsic solubility is defined as the maximum amount of solute dissolved in a given solvent under standard conditions of temperature, pressure and pH. It is a static property. Dissolution rate is defined as the amount of solid substance that goes into solution per unit time under standard conditions of temperature, pH and solvent composition and constant solid surface area. It is a dynamic process. Several drugs have poor aqueous solubility to have a bearing on dissolution rate. The matter is of great concern when the solubility is less than 1 to 2 mg/ml in the pH range of 2 to 8. However, there are well known examples of drugs such as cisapride which despite their low aqueous solubility have sufficient oral bioavailability. Two reasons can be attributed to this—one, the rapid rate of dissolution despite low intrinsic solubility and two, the therapeutic dose of drug may be so small that the GI transit time is sufficient for adequate dissolution and absorption to occur. Thus, in contrast to absolute solubility, the dynamic process of drug dissolution is better related to drug absorption and bioavailability.
Theories of Drug Dissolution
Dissolution is a process in which a solid substance solubilises in a given solvent i.e. mass transfer from the solid surface to the liquid phase. Several theories to explain drug dissolution have been proposed. Some of the important ones are:
1. Diffusion layer model/Film theory,
2. Danckwert’s model/Penetration or Surface renewal theory, and
3. Interfacial barrier model/Double-barrier or Limited solvation theory.
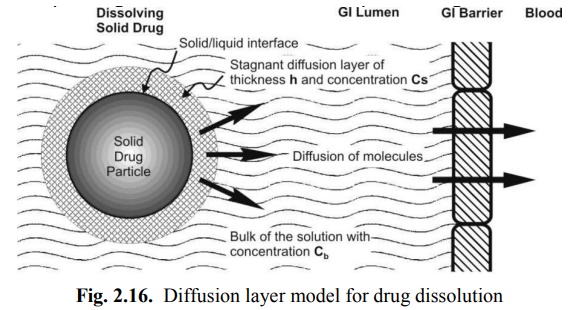
Diffusion Layer Model/Film Theory
This is the simplest and the most common theory for dissolution. Here, the process of dissolution of solid particles in a liquid, in the absence of reactive or chemical forces, consists of two consecutive steps:
1. Solution of the solid to form a thin film or layer at the solid/liquid interface called as the stagnant film or diffusion layer which is saturated with the drug; this step is usually rapid, and
2. Diffusion of the soluble solute from the stagnant layer to the bulk of the solution; this step is slower and is therefore the rate-determining step in drug dissolution. The model is depicted in Fig. 2.16.
The earliest equation to explain the rate of dissolution when the process is diffusion controlled and involves no chemical reaction was given by Noyes and Whitney:

where,
dC/dt = dissolution rate of the drug,
k = dissolution rate constant (first order),
Cs = concentration of drug in the stagnant layer (also called as the saturation or maximum drug solubility), and
Cb = concentration of drug in the bulk of the solution at time t.
Equation 2.3 was based on Fick's second law of diffusion. Nernst and Brunner incorporated Fick‘s first law of diffusion and modified the Noyes-Whitney’s equation to:
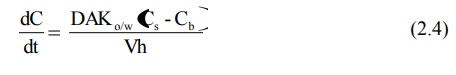
where,
D = diffusion coefficient (diffusivity) of the drug
A= surface area of the dissolving solid
Kw/o = water/oil partition coefficient of the drug considering the fact that dissolution body fluids are aqueous. Since the rapidity with which a drug dissolves depends on the Kw/o, it is also called as the intrinsic dissolution rate constant. It is a characteristic of drugs.
V = volume of dissolution medium.
h= thickness of the stagnant layer.
(Cs – Cb) = concentration gradient for diffusion of drug.
The influence of various parameters in equation 2.4 on drug dissolution is depicted in Table 2.4.
TABLE 2.4
Influence of Some Parameters on Dissolution Rate of Drug
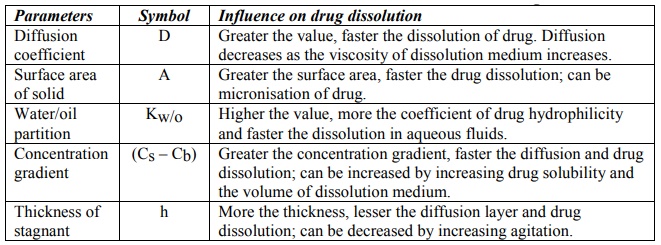
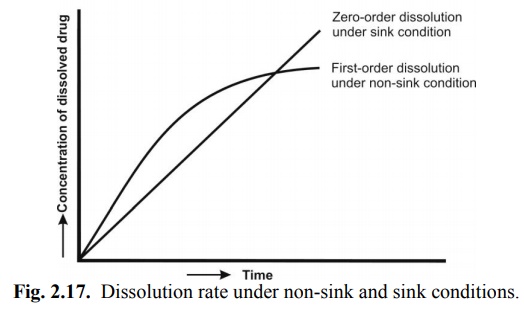
Equation 2.4 represents first-order dissolution rate process, the driving force for which is the concentration gradient (Cs – Cb). Under such a situation, dissolution is said to be under non-sink conditions. This is true in case of in vitro dissolution in a limited dissolution medium. Dissolution in such a situation slows down after sometime due to build-up in the concentration of drug in the bulk of the solution. The in vivo dissolution is always rapid than in vitro dissolution because the moment the drug dissolves; it is absorbed into the systemic circulation. As a result, Cb = 0, and dissolution is at its maximum. Thus, under in vivo conditions, there is no concentration build-up in the bulk of the solution and hence no retarding effect on the dissolution rate of the drug i.e. Cs >> Cb and sink conditions are maintained. Under sink conditions, if the volume and surface area of solid are kept constant, then equation 2.4 reduces to:
dC/dt = K (2.5)

where K incorporates all the constants in equation 2.4. Equation 2.5 represents that the dissolution rate is constant under sink conditions and follows zero-order kinetics i.e. yields a linear plot (Fig. 2.17).
To obtain good in vitro-in vivo dissolution rate correlation, the in vitro dissolution must always be carried under sink conditions. This can be achieved in one or more of the following ways:
1. Bathing the dissolving solid in fresh solvent from time to time.
2. Increasing the volume of dissolution fluid.
3. Removing the dissolved drug by partitioning it from the aqueous phase of the dissolution fluid into an organic phase placed either above or below the dissolution fluid—for example, hexane or chloroform.
4. Adding a water miscible solvent such as alcohol to the dissolution fluid, or
5. By adding selected adsorbents to remove the dissolved drug.
The in vitro sink conditions are so maintained that Cb is always less than 10% of Cs.
The Noyes-Whitney’s equation assumes that the surface area of the dissolving solid remains constant during dissolution, which is practically not possible for dissolving particles. Hence, dissolution methods that involve use of constant surface area discs are employed to determine the rate of dissolution.
To account for the particle size decrease and change in surface area accompanying dissolution, Hixson and Crowell’s cubic root law of dissolution is used:
Wo1/3 – W1/3 = Kt (2.6)
where,
Wo = original mass of the drug
W = mass of the drug remaining to dissolve at time t
K= dissolution rate constant
Danckwert’s Model (Penetration or Surface Renewal Theory)
Danckwert did not approve of the existence of a stagnant layer and suggested that turbulence in the dissolution medium exists at the solid/liquid interface. As a result, the agitated fluid consisting of macroscopic mass of eddies or packets reach the solid/liquid interface in a random fashion due to eddy currents, absorb the solute by diffusion and carry it to the bulk of the solution. Such solute containing packets are continuously replaced with new packets of fresh solvent due to which the drug concentration at the solid/liquid interface never reaches Cs and has a lower limiting value of Ci. Since the solvent packets are exposed to new solid surface each time, the theory is called as surface renewal theory.
The Danckwert’s model is expressed by equation:
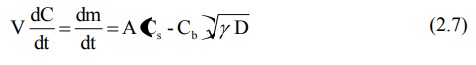
where,
m = mass of solid dissolved, and
γ = rate of surface renewal (or the interfacial tension).
The model is depicted in Fig. 2.18.
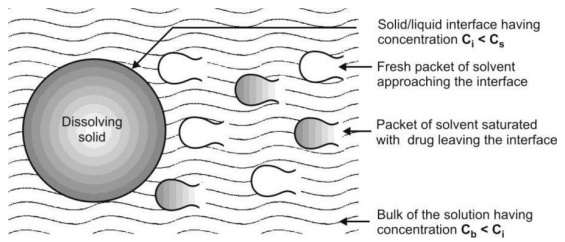
Fig. 2.18. Danckwert’s model for drug dissolution
Interfacial Barrier Model (Double Barrier or Limited Solvation Theory)
The diffusion layer model and the Danckwert’s model were based on two assumptions:
1. The rate-determining step that controls dissolution is the mass transport.
2. Solid-solution equilibrium is achieved at the solid/liquid interface.
According to the interfacial barrier model, an intermediate concentration can exist at the interface as a result of solvation mechanism and is a function of solubility rather than diffusion. When considering the dissolution of a crystal, each face of the crystal will have a different interfacial barrier. Such a concept is given by the following equation:

where,
G = dissolution rate per unit area, and
Ki = effective interfacial transport constant.
In this theory, the diffusivity D may not be independent of saturation concentration Cs. The interfacial barrier model can be extended to both diffusion layer model and the Danckwert’s model (for in vitro drug dissolution models refer chapter 11).
Factors Affecting Drug Dissolution and Dissolution Rate
Factors of in vivo importance that can affect dissolution and hence absorption can be categorized into 2 classes:
1. Physicochemical properties of the drug, and
2. Dosage form factors.
The various physicochemical properties of drug that affect drug dissolution and its rate are—solubility, particle size, polymorphism, salt form, pseudopolymorphism, complexation, wettability, etc. Dosage form factors include several formulation factors and excipients incorporated in the dosage form. Each of these factors will be discussed in detail in the latter part of this chapter.
Of the various factors listed above, the factor of prime importance is drug solubility. Almost every factor that affects dissolution rate, influences the drug solubility in one way or the other. From several equations pertaining to dissolution rate, it is clear that it is directly related to drug solubility. An empirical relation which is useful to predict the dissolution rate of a drug from its solubility is:

where R = dissolution rate of the drug.
It has been shown that a drug should have a minimum aqueous solubility of 1% to avoid bioavailability problems.
2. Particle Size and Effective Surface Area of the Drug
Particle size and surface area of a solid drug are inversely related to each other. Smaller the drug particle, greater the surface area. Two types of surface area of interest can be defined:
1. Absolute surface area which is the total area of solid surface of any particle, and
2. Effective surface area which is the area of solid surface exposed to the dissolution medium.
From the modified Noyes-Whitney equation 2.4, it is clear that larger the surface area, higher the dissolution rate. Since the surface area increases with decreasing particle size, a decrease in particle size, which can be accomplished by micronisation, will result in higher dissolution rates. However, it is important to note that it is not the absolute surface area but the effective surface area that is proportional to the dissolution rate. Greater the effective surface area, more intimate the contact between the solid surface and the aqueous solvent and faster the dissolution. But it is only when micronisation reduces the size of particles below 0.1 microns that there is an increase in the intrinsic solubility and dissolution rate of the drug. The surface of such small particles has energy higher than the bulk of the solid resulting in an increased interaction with the solvent. This is particularly true in case of drugs which are non-hydrophobic, for example, micronisation of poorly aqueous soluble drugs like griseofulvin, chloramphenicol and several salts of tetracycline results in superior dissolution rates in comparison to the simple milled form of these drugs.
Micronisation has in fact enabled the formulator to decrease the dose of certain drugs because of increased absorption efficiency—for example, the griseofulvin dose was reduced to half and that of spironolactone was decreased 20 times following micronisation. However, in case of hydrophobic drugs like aspirin, phenacetin and phenobarbital, micronisation actually results in a decrease in the effective surface area of such powders and thus, a fall in the dissolution rate. Three reasons have been suggested for such an outcome —
1. The hydrophobic surface of the drug adsorbs air onto their surface which inhibit their wettability.
2. The particles re-aggregate to form larger particles due to their high surface free energy, which either float on the surface or settle at the bottom of the dissolution medium.
3. Electrically induced agglomeration owing to surface charges prevents intimate contact of the drug with the dissolution medium.
The net result of these effects is that there is a decrease in the effective surface area available to the dissolution medium and therefore a fall in the dissolution rate.
The absolute surface area of hydrophobic drugs can be converted to their effective surface area by:
1. Use of surfactant as a wetting agent that -
• Decreases the interfacial tension, and
• Displaces the adsorbed air with the solvent.
For example, polysorbate 80 increases the bioavailability of phenacetin by promoting its wettability.
2. Adding hydrophilic diluents such as PEG, PVP, dextrose, etc. which coat the surface of hydrophobic drug particles and render them hydrophilic.
Particle size reduction and subsequent increase in the surface area and dissolution rate is not advisable under following circumstances –
• When the drugs are unstable and degrade in solution form (penicillin G and erythromycin),
• When drugs produce undesirable effects (gastric irritation caused by nitrofurantoin)
• When a sustained effect is desired.
In addition to increasing the dissolution rate, the second mechanism by which a reduction in particle size improves drug dissolution is through an increase in its solubility. However, such an effect can only be achieved by reducing the particle size to a submicron level which is possible by use of one of the following specialized techniques such as formation of:
1. Molecular dispersion/solid solution where the sparingly soluble drug is molecularly entrapped in the lattice of a hydrophilic agent such as cyclodextrins.
2. Solid dispersion where the drug is dispersed in a soluble carrier such as PVP, PEG, urea, etc.
(Refer chapter 11 for methods used in enhancing the bioavailability of drugs).
3. Polymorphism and Amorphism
Depending upon the internal structure, a solid can exist either in a crystalline or amorphous form (Fig. 2.19). When a substance exists in more than one crystalline form, the different forms are designated as polymorphs and the phenomenon as polymorphism. Polymorphs are of two types:
1. Enantiotropic polymorph is the one which can be reversibly changed into another form by altering the temperature or pressure e.g. sulphur, and
2. Monotropic polymorph is the one which is unstable at all temperatures and pressures e.g. glyceryl stearates.
The polymorphs differ from each other with respect to their physical properties such as solubility, melting point, density, hardness and compression characteristics. They can be prepared by crystallizing the drug from different solvents under diverse conditions. The existence of the polymorphs can be determined by using techniques such as optical crystallography, X-ray diffraction, differential scanning calorimetry, etc.
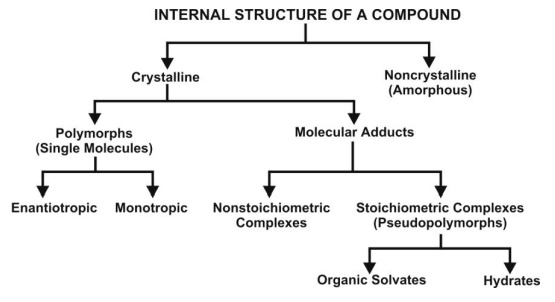
Fig. 2.19. Classification of internal structure of a compound
Depending on their relative stability, one of the several polymorphic forms will be physically more stable than the others. Such a stable polymorph represents the lowest energy state, has highest melting point and least aqueous solubility. The remaining polymorphs are called as metastable forms which represent the higher energy state, have lower melting points and higher aqueous solubilities. Because of their higher energy state, the metastable forms have a thermodynamic tendency to convert to the stable form. A metastable form cannot be called unstable because if it is kept dry, it will remain stable for years.
Since the metastable forms have greater aqueous solubility, they show better bioavailability and are therefore preferred in formulations—for example, of the three polymorphic forms of chloramphenicol palmitate -A, B and C, the B form shows best availability and the A form is virtually inactive biologically. The polymorphic form III of riboflavin is 20 times more water-soluble than the form I. Only 10% of the pharmaceuticals are present in their metastable forms. However, because of their poor thermodynamic stability, aging of dosage forms containing such metastable forms usually result in formation of less soluble, stable polymorph—for example, the more soluble crystalline form II of cortisone acetate converts to the less soluble form V in an aqueous suspension resulting in caking of solid. Such a transformation of metastable to stable form can be inhibited by dehydrating the molecule environment or by adding viscosity building macromolecules such as PVP, CMC, pectin or gelatin that prevent such a conversion by adsorbing onto the surface of the crystals.
About 40% of all organic compounds can exist in various polymorphic forms. Seventy percent of the barbiturates and 65% of sulphonamides exhibit polymorphism. Barbital, methyl paraben and sulphapyridine can exist in as many as 6 polymorphic forms and cortisone acetate in 8 forms.
Some drugs can exist in amorphous form (i.e. having no internal crystal structure). Such drugs represent the highest energy state and can be considered as supercooled liquids. They have greater aqueous solubility than the crystalline forms because the energy required to transfer a molecule from crystal lattice is greater than that required for non-crystalline (amorphous) solid—for example, the amorphous form of novobiocin is 10 times more soluble than the crystalline form. Chloramphenicol palmitate, cortisone acetate and phenobarbital are other examples where the amorphous forms exhibit higher water solubility. Thus, the order for dissolution of different solid forms of drugs is —
Amorphous > Metastable > Stable.
4. Hydrates/Solvates (Pseudopolymorphism)
The crystalline form of a drug can either be a polymorph or a molecular adduct or both. The stoichiometric type of adducts where the solvent molecules are incorporated in the crystal lattice of the solid are called as the solvates, and the trapped solvent as solvent of crystallization. The solvates can exist in different crystalline forms called as pseudopolymorphs. This phenomenon is called as pseudopolymorphism. When the solvent in association with the drug is water, the solvate is known as a hydrate. Hydrates are most common solvate forms of drugs.
Generally, the anhydrous form of a drug has greater aqueous solubility than the hydrates. This is because the hydrates are already in interaction with water and therefore have less energy for crystal break-up in comparison to the anhydrates (thermodynamically higher energy state) for further interaction with water. The anhydrous form of theophylline and ampicillin have higher aqueous solubilities, dissolve at a faster rate and show better bioavailability in comparison to their monohydrate and trihydrate forms respectively. On the other hand, the organic (nonaqueous) solvates have greater aqueous solubility than the non-solvates—for example, the n-pentanol solvate of fludrocortisone and succinylsulphathiazole and the chloroform solvate of griseofulvin are more water-soluble than their non-solvated forms. Like polymorphs, the solvates too differ from each other in terms of their physical properties. In case of organic solvates, if the solvent is toxic, they are not of therapeutic use.
5. Salt Form of the Drug
Most drugs are either weak acids or weak bases. One of the easiest approaches to enhance the solubility and dissolution rate of such drugs is to convert them into their salt forms. Generally, with weakly acidic drugs, a strong base salt is prepared such as the sodium and potassium salts of barbiturates and sulphonamides. In case of weakly basic drugs, a strong acid salt is prepared like the hydrochloride or sulphate salts of several alkaloidal drugs.
At a given pH, the solubility of a drug, whether acidic/basic or its salt form is a constant. The influence of salt formation on the drug solubility, rate of dissolution and absorption can be explained by considering the pH of the diffusion layer and not the pH of the bulk of the solution (refer diffusion layer theory of drug dissolution). Consider the case of a salt of a weak acid. At any given pH of the bulk of the solution, the pH of the diffusion layer (saturation solubility of the drug) of the salt form of a weak acid will be higher than that observable with the free acid form of the drug (can be practically observed in the laboratory). Owing to the increased pH of the diffusion layer, the solubility and dissolution rate of a weak acid in this layer is promoted; since it is a known fact that higher pH favours the dissolution of weak acids. Thus, if dissolution is faster, absorption is bound to be rapid. In case of salts of weak bases, the pH of the diffusion layer will be lower in comparison to that found with the free base form of the drug. Consequently, the solubility of a basic drug at this lower pH is enhanced. Thus, if:
[H+]d = hydrogen ion concentration of the diffusion layer, and
[H+]b = hydrogen ion concentration of the bulk of the solution, then,
for salts of weak acids, [H+]d < [H+]b
for salts of weak bases, [H+]d > [H+]b
The increase and decrease in pH of the diffusion layer by the salts of weak acids and bases have been attributed to the buffering action of strong base cation and strong acid anion respectively.
Yet another convincing reason for enhanced solubility of salts of weak acids is the precipitation of the drug as very fine particles. When the soluble ionic form of the drug diffuses from the stagnant diffusion layer into the bulk of the solution whose pH is low, it is transformed into its free acid form having lesser aqueous solubility at the lower pH of the bulk solution. Consequently, this free acidic form of the drug is precipitated in the form of fine particles. The resultant increase in the surface area is then responsible for the rapid dissolution and absorption in comparison to the drug administered in just the acidic form (Fig. 2.20).
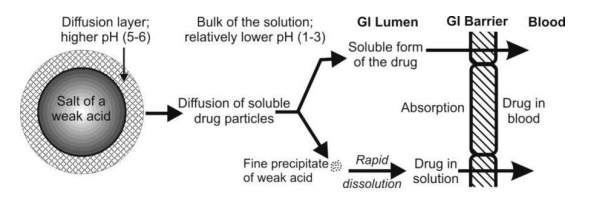
Fig. 2.20. Dissolution and absorption of an acidic drug administered in a salt form
The principle of in situ salt formation has been utilized to enhance the dissolution and absorption rate of certain drugs like aspirin and penicillin from buffered alkaline tablets. The approach is to increase the pH of the microenvironment of the drug by incorporating buffer agents and promote dissolution rate. Apart from the enhanced bioavailability, buffered aspirin tablets have two more advantages: firstly, the gastric irritation and ulcerogenic tendency of the drug is greatly reduced, and secondly, the problem with the use of sodium salt of aspirin (to enhance the solubility) which otherwise has poor hydrolytic stability, is overcome by in situ salt formation.
The selection of appropriate salt form for better dissolution rate is also important. It has been shown that the choline and the isopropanolamine salts of theophylline dissolve 3 to 4 times more rapidly than the ethylenediamine salt and show better bioavailability.
A factor that influences the solubility of salt forms of the drug is the size of the counter ion. Generally speaking, smaller the size of the counter ion, greater the solubility of salt—for example, the bioavailability of novobiocin from its sodium salt, calcium salt and free acid form was found to be in the ratio — 50 : 25 : 1. Where the counter ion is very large in size and/or has poor ionic strength (as in the case of ester form of drugs), the solubility may be much lower than the free drug itself—for example, the pamoates, stearates and palmitates of weak bases have poor aqueous solubility. These forms are, however, useful in several ways such as to prolong the duration of action (steroidal salts), to overcome bad taste (chloramphenicol palmitate), to enhance GI stability (erythromycin estolate) or to decrease the side effects, local or systemic.
There are exceptions where the so called more soluble salt form of the drug showed poor bioavailability. One such study was the comparative dissolution of sodium phenobarbital and free phenobarbital from their tablets. Slower dissolution with sodium salt was observed and the reason attributed to it was that its tablet swelled but did not disintegrate and thus dissolved slowly. An identical result was obtained with hydrochloride salts of several tetracycline analogs and papaverine; better dissolution and bioavailability was observed with the free bases. The reason for poor solubility and dissolution rate was the suppression action of the common ion effect.
6. Drug pKa and Lipophilicity and GI pH—pH Partition Hypothesis
The pH partition theory (Brodie et al) explains in simple terms, the process of drug absorption from the GIT and its distribution across all biological membranes. The theory states that for drug compounds of molecular weight greater than 100, which are primarily transported across the biomembrane by passive diffusion, the process of absorption is governed by:
1. The dissociation constant (pKa) of the drug.
2. The lipid solubility of the unionised drug (a function of drug Ko/w).
3. The pH at the absorption site.
Since most drugs are weak electrolytes (weak acids or weak bases), their degree of ionisation depends upon the pH of the biological fluid. If the pH on either side on the membrane is different, then the compartment whose pH favours greater ionisation of the drug will contain greater amount of drug, and only the unionised or undissociated fraction of drug, if sufficiently lipid soluble, can permeate the membrane passively until the concentration of unionised drug on either side of the membrane becomes equal i.e. until equilibrium is attained.
The above statement of the hypothesis was based on the assumptions that:
1. The GIT is a simple lipoidal barrier to the transport of drug.
2. Larger the fraction of unionised drug, faster the absorption.
3. Greater the lipophilicity (Ko/w) of the unionised drug, better the absorption.
7. Drug pKa and Gastrointestinal pH
The amount of drug that exists in unionised form is a function of dissociation constant (pKa) of the drug and pH of the fluid at the absorption site.
It is customary to express the dissociation constants of both acidic and basic drugs by pKa values. The lower the pKa of an acidic drug, stronger the acid i.e. greater the proportion of ionised form at a particular pH. Higher the pKa of a basic drug, stronger the base. Thus, from the knowledge of pKa of drug and pH at the absorption site (or biological fluid), the relative amount of ionised and unionised drug in solution at a particular pH and the percent of drug ionised at this pH can be determined by Henderson-Hasselbach equations:
for weak acids,

When the concentration of ionised and unionised drug becomes equal, the second term of equations 2.10 and 2.12 reduces to zero (since log 1 = zero), and thus pH = pKa. The pKa is a characteristic of the drug.
If there is a membrane barrier that separates the aqueous solutions of different pH such as the GIT and the plasma, then the theoretical ratio R of drug concentration on either side of the membrane can be given by equations derived by Shore et al:
for weak acids, for weak bases,

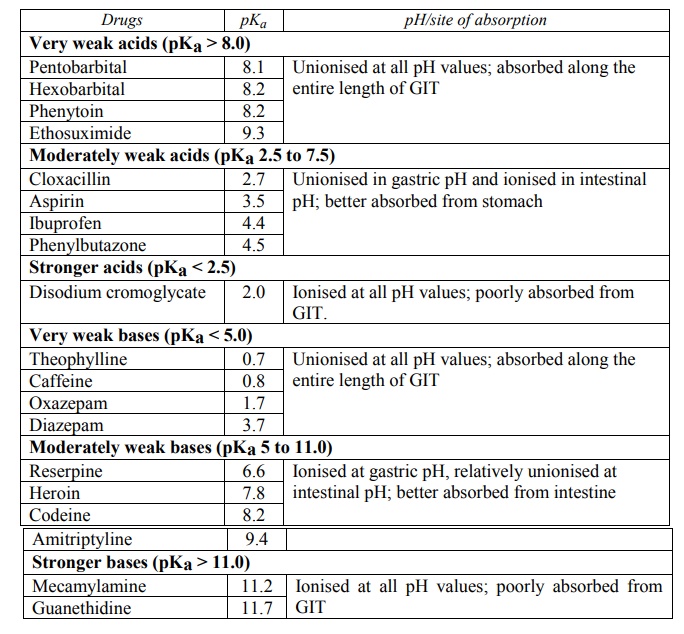
If one considers the pH range in the GIT from 1 to 8, that of the stomach from 1 to 3 and of the intestine (from duodenum to colon) 5 to 8, then certain generalisations regarding ionisation and absorption of drugs can be made, as predicted from the pH-partition hypothesis:
For Weak Acids:
1. Very weak acids (pKa > 8) such as phenytoin, ethosuximide and several barbiturates are essentially unionised at all pH values and therefore their absorption is rapid and independent of GI pH.
2. Acids in the pKa range 2.5 to 7.5 are greatly affected by changes in pH and therefore their absorption is pH-dependent; e.g. several NSAIDs like aspirin, ibuprofen, phenylbutazone, and a number of penicillin analogs. Such drugs are better absorbed from acidic conditions of stomach (pH < pKa) where they largely exist in unionised form.
3. Stronger acids with pKa < 2.5 such as cromolyn sodium are ionised in the entire pH range of GIT and therefore remain poorly absorbed.
For Basic Drugs:
1. Very weak bases (pKa < 5.0) such as caffeine, theophylline and a number of benzodiazepines like diazepam, oxazepam and nitrazepam are essentially unionised at all pH values and therefore their absorption is rapid and pH-independent.
2. Bases in the pKa range 5 to 11.0 are greatly affected by changes in pH and hence their absorption is pH-dependent; e.g. several morphine analogs, chloroquine, imipramine and amitriptyline. Such drugs are better absorbed from the relatively alkaline conditions of the intestine where they largely exist in unionised form.
3. Stronger bases with pKa > 11.0 like mecamylamine and guanethidine are ionised in the entire pH range of GIT and therefore poorly absorbed.
A summary of above discussion is given in Table 2.5.
TABLE 2.5.
Influence of drug pKa and GI pH on Drug Absorption
By using equations from 2.10 to 2.15, one can calculate the relative amounts of unionised (absorbable) and ionised (unabsorbable) forms of the drug and predict the extent of absorption at a given pH of GIT. An example of this is illustrated in figure 2.21.

Fig. 2.21. Influence of pH on ionisation of drug. [HA] and [BOH] are concentration of unionised acid and base, and [A-] and [B+] are concentration of ionised acid and base respectively
Besides the dissociation constant pKa, total aqueous solubility, ST, of an ionisable drug is an important factor in the passive absorption of drugs. It is defined as the sum of concentration of ionised drug in solution and concentration of unionised drug in solution. The solubility of unionised form of the drug is known as the intrinsic solubility of the drug. If Sa is the intrinsic solubility of weakly acidic drugs and Sb that of weakly basic drugs, then –
for acidic drugs,
ST = Sa [1 + 10(pH –pKa)] (2.16)
for basic drugs,
ST = Sb [1 + 10(pKa –pH)] (2.17)
Fig. 2.22 illustrates pH-solubility profile for a free acid and free base of weakly acidic and weakly basic drugs.
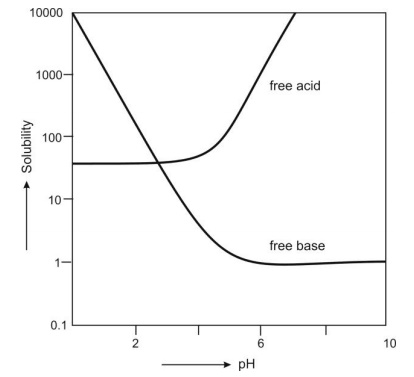
Fig. 2.22 pH-solubility curve for weakly acidic and weakly basic drugs
Certain conclusions and generalizations can now be stated – For weakly acidic drugs,
1. When pH > pKa, ST >> Sa because ionisation of drug increases tremendously.
2. When pH = pKa, ST = 2Sa, because the drug is 50% ionised.
3. When pH < pKa, ST ≡ Sa since the drug exists predominantly in unionised form.
For weakly basic drugs,
1. When pH > pKa, ST ≡ Sb since the drug exists predominantly in unionised form.
2. When pH = pKa, ST = 2Sb, because the drug is 50% ionised.
3. When pH < pKa, ST >> Sb because ionisation of drug increases tremendously.
Lipophilicity and Drug Absorption
As mentioned earlier, it is the pKa of a drug that determines the degree of ionisation at a particular pH and that only the unionised drug, if sufficiently lipid soluble, is absorbed into the systemic circulation. Thus, even if the drug exists in the unionised form, it will be poorly absorbed if it has poor lipid solubility (or low Ko/w). Ideally, for optimum absorption, a drug should have sufficient aqueous solubility to dissolve in the fluids at the absorption site and lipid solubility (Ko/w) high enough to facilitate the partitioning of the drug in the lipoidal biomembrane and into the systemic circulation. In other words, a perfect hydrophilic-lipophilic balance (HLB) should be there in the structure of the drug for optimum bioavailability.
The lipid solubility of a drug is measured by a parameter called as log P where P is oil/water partition coefficient (Ko/w or simply P) value of the drug. This value is a measure of the degree of distribution of drug between lipophilic solvents such as n-octanol and an aqueous phase (water or a suitable buffer). In general, the octanol/pH 7.4 buffer partition coefficient value in the range of 1 to 2 of a drug is sufficient for passive absorption across lipoidal membranes. A direct correlation between a drug’s Ko/w and extent of absorption is illustrated in Table 2.6.
TABLE 2.6.
Comparison between Intestinal Absorption of Some Drugs through the Rat Intestine and Ko/w of the Ionised Form of the Drugs
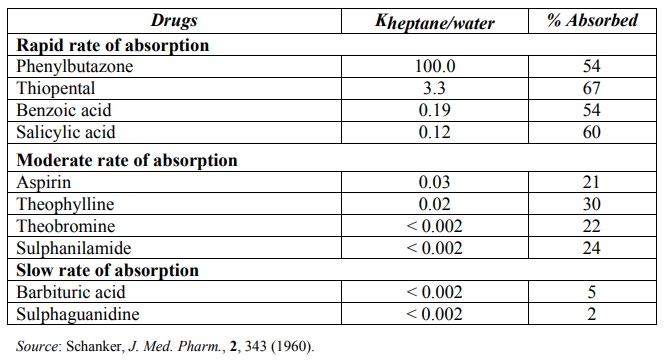
For ionisable drugs where the ionised species does not partition into the aqueous phase, the apparent partition coefficient (D) can be calculated from following equations –
for acidic drugs,
log D = log P - log [1 + 10(pH –pKa)] (2.18)
for basic drugs,
log D = log P - log [1 + 10(pKa –pH)] (2.19)
Limitations of pH-Partition Hypothesis
The pH-partition hypothesis over-simplified the otherwise complicated process of drug absorption and therefore has its own limitations. Some of the deviations from the theory are:
1. Presence of virtual membrane pH
2. Absorption of ionised drug
3. Influence of GI surface area and residence time of drug
4. Presence of aqueous unstirred diffusion layer
1. Presence of Virtual Membrane pH: The pH-partition hypothesis suggested that only the unionised drug at a given GI lumen pH is absorbed. An S-shaped curve, called as the pH-absorption curve denoting the dissociation of drug, is obtained when pH is plotted versus rate of drug absorption (Fig. 2.23).
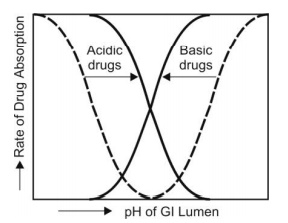
Fig. 2.23. pH-absorption curve for acidic and basic drugs. Dotted lines indicate curves predicted by pH-partition hypothesis and bold lines indicate the practical curves.
However, differences in the extent of absorption of salicylic acid has been observed at a given GI pH than that predicted by pH-partition hypothesis. The experimental pH-absorption curves are less steep and shift to the left (lower pH values) for a basic drug and to the right (higher pH values) for an acidic drug. This led to the suggestion that a virtual pH, also called as the microclimate pH, different from the luminal pH exists at the membrane surface. This virtual membrane pH actually determines the extent of drug ionisation and thus, drug absorption.
2. Absorption of Ionised Drugs: An important assumption of the theory was that only unionised form of the drug is absorbed and permeation of the ionised drug is negligible since its rate of absorption is 3 to 4 times less than that of unionised drug. This is called as principle of non-ionic diffusion. The principle is true to a large extent as ionised drugs have low lipid solubility and relatively poor permeability. However, the pH-absorption curve shift suggested that ionised forms of some drugs also get absorbed to a considerable extent. If such drugs have a large lipophilic group in their structure, despite their ionisation, they will be absorbed passively—for example, morphinan derivatives. Other mechanisms are also involved in the absorption of ionised drugs such as active transport, ion-pair transport and convective flow.
3. Influence of GI Surface Area and Residence Time of Drug: According to the pH-partition theory, acidic drugs are best absorbed from stomach (acidic pH) and basic drugs from intestine (alkaline pH) in which conditions they are unionised to a large extent. This could be true under conditions where the surface area of stomach and intestine are same. It could also mean that once an acidic drug reaches the intestine, the remaining fraction will be poorly absorbed and that unless a basic drug reaches the intestine and gets absorbed considerably, it may not be able to attain its therapeutic level. But, irrespective of the GI pH and the degree of ionisation, both acidic and basic drugs are more rapidly absorbed from the intestine, primarily because of its large surface area and secondly, because of long residence time of the drug in the intestine.
4. Presence of Aqueous Unstirred Diffusion Layer: The pH-shift in the absorption of acidic and basic drugs, as discussed earlier, also accounts for the fact that the bulk of the luminal fluid is not in direct contact with the membrane but a barrier called as aqueous unstirred diffusion layer is interposed between them. Such a model is depicted in Fig. 2.24.
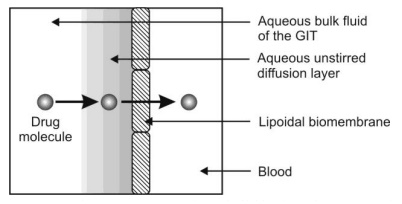
Fig. 2.24. Presence of aqueous unstirred diffusion layer on the membrane surface
Such a layer has a real thickness and is a barrier to absorption of drugs. In the original pH-partition theory, the rate-limiting step in the absorption of drugs was the partitioning in the lipid barrier. With the incorporation of unstirred aqueous diffusion layer, a drug must diffuse first through this aqueous barrier and then through the lipoidal barrier. Thus, drugs having large partition coefficient can rapidly penetrate the lipid membrane but diffusion through unstirred water layer is the rate-limiting step in their absorption. This applies in particular to high molecular weight fatty acids and bile acids.
Despite its limitations, the pH-partition theory is still useful in the basic understanding of drug absorption and movement of drug between various body compartments.
Drug Permeability and Absorption
Most orally administered drugs enter the systemic circulation by passive diffusion and their absorption is expressed mathematically by equation –
M = Peff A Capp tres (2.20)
where,
M = amount of drug absorbed
Peff = effective membrane permeability
A = surface area available for absorption
Capp = apparent luminal drug concentration
tres = residence time of drug in GI lumen.
Since it is difficult to alter or control surface area and residence time of drug, focus for promoting absorption depends on enhancing permeability of drug or drug concentration at absorption site (see BCS classification of drugs, Table 2.3).
The three major drug characteristics that determine the passive transport or permeability of drugs across intestinal epithelium are –
· Lipophilicity of drug expressed as log P.
· Polarity of drug which is measured by the number of H-bond acceptors and number of H-bond donors on the drug molecule.
· Molecular size.
The net effect of the above three properties of drug on its permeability across intestinal epithelium is given as Rule of Five by Lipinski et al which is written as –
· Molecular weight of drug ≤ 500
· Lipophilicity of drug, log P ≤ 5
· Number of H-bond acceptors ≤ 10
· Number of H-bond donors ≤ 5
For a given drug, if any two of these values is greater than that specified above, then oral absorption may be significant problem.
8. Drug Stability
A drug for oral use may destabilize either during its shelf-life or in the GIT. Two major stability problems resulting in poor bioavailability of an orally administered drug are—degradation of the drug into inactive form, and interaction with one or more different component(s) either of the dosage form or those present in the GIT to form a complex that is poorly soluble or is unabsorbable. Destabilization of a drug during its shelf-life and in the GIT will be discussed in detail under formulation factors and patient related factors respectively.
Stereochemical Nature of Drug
Chiral drugs constitute approximately 60% of the drugs in current use. Majority of these are marketed as racemic mixtures. Although it is well established that optical isomers differ in the potency of pharmacological effect, it is only recently that attention is being paid to influence of chirality on pharmacokinetic processes like absorption, distribution and elimination.
Enantiomers possess identical physical and chemical properties despite significant differences in spatial configuration. Thus, biological processes which are passive in nature (and thereby depend only upon physical and chemical characteristics of the molecule) do not display selectively for one isomer over another. However, biological processes such as protein binding which require interaction of a drug with a macromolecule may exhibit stereoselectivity.
As majority of drugs are absorbed passively, they do not display stereoselectivity. Conversely, demonstration of stereoselective absorption would be strong evidence that a drug is absorbed by a carrier-mediated process.
Related Topics
