Protein synthesis and its selective inhibition
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Mechanisms of action of antibiotics and synthetic anti-infective agents
Bacterial ribosomes are smaller than their mammalian counterparts. They consist of one 30S and one 50S subunit (the S suffix denotes the size, which is derived from the rate of sedimentation in an ultracentrifuge). The 30S subunit comprises a single strand of 16S rRNA and over 20 different proteins that are bound to it.
PROTEIN SYNTHESIS AND ITS SELECTIVE INHIBITION
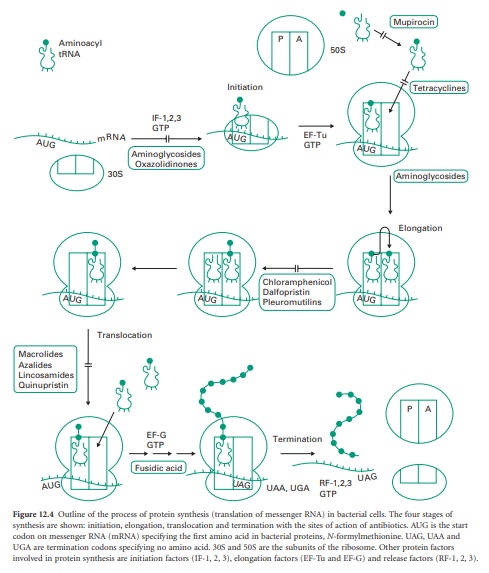
Figure 12.4 outlines
the process of protein synthesis involving the ribosome, mRNA, a series of
aminoacyl transfer RNA (tRNA) molecules (at least one for each amino acid) and
accessory protein factors involved in initiation, elongation and termination.
As the process is essentially the same in prokaryotic (bacterial) and
eukaryotic cells (i.e. higher organisms and mammalian cells) it is surprising
that there are so many selective agents which act in this area (see Figure
12.1).
Figure 12.4 Outline
of the process of protein synthesis (translation of messenger RNA) in bacterial
cells. The four stages of synthesis are shown: initiation, elongation,
translocation and termination with the sites of action of antibiotics. AUG is
the start codon on messenger RNA (mRNA) specifying the first amino acid in
bacterial proteins, N-formyl-methionine. UAG, UAA and UGA are termination
codons specifying no amino acid. 30S and 50S are the subunits of the ribosome.
Other protein factors involved in protein synthesis are initiation factors
(IF-1, 2, 3), elongation factors (EF-Tu and EF-G) and release factors (RF-1, 2,
3).
Bacterial ribosomes are smaller than their mammalian counterparts.
They consist of one 30S and one 50S
subunit (the S suffix
denotes the size, which is derived
from the rate of sedimentation in an ultracentrifuge). The 30S
subunit comprises a single strand
of 16S rRNA and
over 20 different proteins that are bound to it. The
larger 50S subunit contains two single strands
of rRNA (23S and 5S) together
with over 30 different proteins. The subunits pack together to form an intact 70S
ribosome. The equivalent subunits
for mammalian ribosomes are 40S and 60S, making an 80S ribosome. Some agents exploit subtle differences in structure between
the bacterial and mammalian ribosomes. The macrolides, azalides and chloramphenicol act on the 50S subunits in bacteria
but not the 60S subunits of mammalian cells.
By contrast, the tetracyclines derive their selective action
through active uptake and concentration within microbial cells but only limited
penetration of mammalian cells.
Aminoglycoside–aminocyclitol antibiotics
Most of the information on the mechanisms of action of aminoglycoside–aminocyclitol (AGAC) antibiotics comes from studies
with streptomycin. One effect of AGACs is
to interfere with the initiation and assembly of the bacterial ribosome (Figure 12.4).
During assembly of the initiation complex, N-formylmethionyl-tRNA (fmet-tRNA) binds initially to the ribosome
binding site on the untranslated 5′ end of the mRNA
together with the 30S
ribosomal subunit. Three protein
initiation factors (designated IF-1, 2 and 3) and a molecule
of guanosine triphosphate (GTP) are involved in positioning the fmet-tRNA on the AUG start
codon of mRNA.
IF-1 and IF-3 are then released from the complex,
GTP is hydrolysed to guanosine diphosphate (GDP) and released with IF-2 as the 50S subunit joins
the 30S subunit
and mRNA to form a functional ribosome. The fmet-tRNA occupies the peptidyl site (P site) leaving
a vacant acceptor
site (A site) to receive the next aminoacyl-tRNA specified by the next codon on the mRNA. Streptomycin binds tightly to one of the protein components of the 30S
subunit. Binding of the antibiotic to the protein,
which is the receptor for IF-3,
prevents initiation and assembly of the
ribosome.
Streptomycin binding
to the 30S subunit also distorts
the shape
of the A site on the ribosome and interferes with the positioning of the aminoacyl-tRNA molecules during peptide chain elongation. Streptomycin therefore exerts two effects: inhibition of protein synthesis by freezing the initiation complex, and misreading of the codons through distortion of the
30S subunit. Simple
blockage of protein synthesis would be bacteriostatic rather than bactericidal.
As streptomycin and the other
AGACs exert a potent lethal
action, it seems that the formation of toxic, non-functional proteins through misreading of the codons on mRNA is a more likely
mechanism of action. This can be demonstrated with cell-free translation systems in which
isolated bacterial ribosomes are supplied with artificial mRNA template such as poly(U)
or poly(C) and all the other factors, including aminoacyl-tRNAs needed for protein synthesis.
In the absence of an AGAC the ribosomes will
produce artificial polypeptides, polyphenylalanine (as specified
by the codon
UUU) or polyproline (as specified by the codon CCC). However, when
streptomycin is added,
the ribosomes produce a mixture of polythreonine (codon ACU) and polyserine (codon
UCU). The misreading of the codons
does not appear
to be random: U is read as A
or C, and C is read as A or U. If such misreading occurs in whole cells, the accumulation of non-functional or toxic proteins
would eventually prove fatal to the cells. There is some evidence
that the bacterial cell membrane is damaged
when the cells
attempt to excrete
the faulty proteins.
The effectiveness of the AGACs is enhanced by their
active uptake
by bacteria, which
proceeds in three
phases. First, a rapid uptake occurs within a few seconds of contact, which represents binding of
the positively charged AGAC molecules to the negatively charged surface of the bacteria. This phase is referred to as the energy-independent phase
(EIP) of uptake.
In the case of
Gram-negative bacteria the AGACs damage
the outer membrane
causing release of some lipopolysaccharide, phospholipid
and proteins but this is not directly lethal to the cells.
Second, there follows
an energy-dependent phase of uptake
(EDP I) lasting
about 10 minutes,
in which the AGAC is actively transported across the cytoplasmic membrane. A second
energy-dependent phase (EDP II) which leads
to further intracellular accumulation follows after
some AGAC has bound
to the ribosomes in the cytoplasm. Although the precise
details of uptake by EDP
I and EDP II are
not clear, both require organisms to be growing aerobically. Anaerobes do not take up AGACs by EDP I or EDP II and are consequently
resistant to their action.
Tetracyclines
This group
of antibiotics is actively transported into bacterial cells, possibly as the magnesium complex, achieving
a 50-fold concentration inside the cells. Mammalian
cells do not actively take up the tetracyclines (small
amounts enter by diffusion alone)
and it is this difference in uptake that determines the selective toxicity. Resistance to the tetracyclines occurs through failure of the
active uptake system or the action of active efflux
pumps, which remove the drug from
the cells before
it can interfere with ribosome
function. Other resistance mechanisms involve ribosomal protection and modification. Protein
synthesis by both bacterial and mammalian ribosomes is inhibited
by the tetracyclines in cell-free systems. The action is on
the smaller subunit. Binding of just one molecule of tetracycline to the bacterial 30S subunit occurs
at a site involving the 3′ end of the 16S rRNA, a number of associated ribosomal proteins and magnesium ions. The effect is to block
the binding of aminoacyl-tRNA to the A site
of the ribosome and
halt protein synthesis. Tetracyclines are
bacteriostatic rather than bactericidal, consequently they should not
be used in combination with
β-lactams, which require
cells to be growing and dividing to exert their lethal action.
Chloramphenicol
Of the four possible optical isomers of chloramphenicol, only the d-threo form is active. This antibiotic selectively inhibits protein synthesis in bacterial ribosomes by binding to the 50S subunit in the region of the A site involving the 23S rRNA. The normal binding of the aminocyl-tRNA in the A site is affected
by chloramphenicol in such a way that the peptidyl
transferase cannot form a new peptide bond with the growing peptide chain on the tRNA in the P site. Studies
with aminocyl-tRNA fragments containing truncated tRNA chains
suggest that the shape of the region of tRNA closest
to the amino acid is distorted by chloramphenicol. The altered orientation of this region
of the aminoacyl-tRNA in the A site is sufficient to prevent peptide
bond formation. Chloramphenicol has a broad
spectrum of activity, which covers Gram-positive and Gram-negative bacteria, mycoplasmas, rickettsia and chlamydia. It has the valuable property of penetrating into
mammalian cells and is
therefore the drug of choice
for treatment of intracellular
pathogens, including Salmonella enterica serovar Typhi, the causative organism
of typhoid. Although
it does not inhibit 80S ribosomes, the 70S ribosomes
of mammalian mitochondria are sensitive and therefore some inhibition
occurs in rapidly growing mammalian
cells with high mitochondrial activity.
Macrolides and azalides
Erythromycin is a member
of the macrolide group of antibiotics;
it selectively inhibits
protein synthesis in a
broad range of bacteria by binding to the 50S subunit.
The site at which it binds is close to that of chloramphenicol and involves
the 23S rRNA. Resistance to chloramphenicol and
erythromycin can occur
by methylation of different bases within the same region
of the 23S rRNA. The sites
are therefore not identical, but binding of one
antibiotic prevents binding
of the other. Unlike chloramphenicol, erythromycin blocks translocation.
This is the process by which the ribosome moves along the mRNA
by one codon after the
peptidyl transferase reaction has joined the peptide
chain to the
aminoacyl-tRNA in the
A site. The peptidyl-tRNA is moved (translocated) to the P site, vacating the A site for the next aminocyl-tRNA. Energy is derived by hydrolysis of GTP to GDP by an
associated protein
elongation factor, EF-G.
By blocking the translocation process,
erythromycin causes release
of incomplete polypeptides from the ribosome.
It is assumed that the azalides, such as azithromycin, have a similar
action to the macrolides. The azalides have improved intracellular penetration
over the macrolides and are resistant to the metabolic conversion which reduces the serum half-life of erythromycin.
Clindamycin
This agent
binds selectively to a region
of the 50S ribosomal subunit close
to that of chloramphenicol and erythromycin. It blocks
elongation of the peptide chain
by inhibition of peptidyl transferase.
Streptogramins Quinupristin And Dalfopristin
The two unrelated streptogramins, quinupristin and dalfopristin,
have been used
in combination (in
a 30 : 70 ratio) to treat infections caused by
staphylococci and enterococci, particularly methicillin-resistant Staph. aureus (MRSA) and VRE. Their action
is synergistic, and is
generally bactericidal compared with either agent used alone or compared with antibiotics in the
macrolide group. The
main target is the bacterial 50S ribosome, with
the formulation acting
to inhibit protein synthesis.
The agents bind
sequentially to the 50S subunit; dalfopristin
alters the shape
of the subunit
so that more quinupristin
can bind. Dalfopristin blocks an early step in protein synthesis by forming a bond with
the ribosome, preventing elongation of the
peptide chain by the peptidyl transferase.
Quinupristin blocks a later step by preventing the extension of peptide chains and
causing incomplete chains to be released. The
overall effect is to block
elongation. Use of streptogramins is limited by vasculitis, causing pain on intravenous
administration.
Oxazolidinones—linezolid
Oxazolidinones such
as linezolid act at the early stage
of protein
synthesis, preventing the formation of the initiation complex between the 30S subunit, mRNA and fmet-tRNA.
Mupirocin
The target
of mupirocin is one of a group
of enzymes which couple amino acids to their respective tRNAs for delivery to the ribosome
and incorporation into
protein. The particular enzyme inhibited by mupirocin is involved
in producing isoleucyl-tRNA. The basis
for the inhibition is a structural similarity between one end of the mupirocin molecule and isoleucine. Protein
synthesis is halted when the ribosome encounters the isoleucine
codon through depletion of the pool of isoleucyl-tRNA.
Fusidic acid
This steroidal antibiotic does not act on the ribosome itself, but on one of the associated elongation factors, EF-G. This factor supplies
energy for translocation by hydrolysis of GTP and GDP. Another
elongation factor, EF-Tu, promotes
binding of aminoacyl-tRNA molecules to the A site through
binding and hydrolysis of GTP. Both
EF-G and EF-Tu have overlapping binding
sites on the ribosome. Fusidic acid binds
the EF-G : GDP complex
to the ribosome after
one round of translocation has taken
place. This prevents
further incorporation of aminoacyl-tRNA by blocking
the binding of EF-Tu
: GTP. Fusidic acid owes its selective
antimicrobial action to active
uptake by bacteria and exclusion from mammalian cells. The equivalent elongation factor in mammalian cells, EF-2, is susceptible to fusidic acid
in cell-free systems.
Pleuromutilins - Retapumilins
These agents
bind to the 23S rRNA component of the 50S bacterial ribosome and block
peptide bond formation by interfering with the binding
of the peptidyl transferase
region with the aminoacyl-tRNA substrates in the A and
P sites on the ribosome. This mechanism is different to that
of other peptidyl
transferase inhibitors (chloramphenicol and clindamycin) so cross-resistance to these
agents does not occur.
Related Topics
