Sterility Testing: Pharmaceutical Products
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Sterility Testing: Pharmaceutical Products
A sterility test may be defined as - ‘a test that critically assesses whether a sterilized pharmaceutical product is free from contaminating microorganisms’.
STERILITY
TESTING : PHARMACEUTICAL PRODUCTS
INTRODUCTION
A sterility test may be defined as - ‘a test that critically assesses whether a
sterilized pharmaceutical product is free from contaminating microorganisms’.
According
to Indian Pharmacopoea (1996) the sterility testings are intended for
detecting the presence of viable forms of microorganisms in or on the
pharmacopoeal preparations.
In actual
practice, one invariably comes across certain absolutely important guidelines
and vital precautionary measures that must be adhered to strictly so as to
accomplish the utmost accuracy and precision of the entire concept of sterility
testing for life-saving secondary pharmaceutical products (drugs). A few such cardinal factors, guidelines, and necessary
details are as enumerated under :
(a) Sterility testing, due to
its inherent nature, is intimately associated with a statistical process
wherein the portion of a batch is sampled almost randomly* ; and, therefore,
the chance of the particular batch (lot) duly passed for actual usage
(consumption) solely depends upon the ‘sample’
having passed the stringent sterility test.
(b) Sterility tests should
be performed under conditions designed to avoid accidental contamination of the
product (under investigation) during the test. Nevertheless, such particular
precautions precisely taken for this purpose must not, in any case, adversely
affect any microbes that should be revealed in the test ultimately.
(c) Working
environment wherein the sterility tests
are meticulously carried out must be adequately monitored at regular intervals
by sampling the air and the surface of the working area by performing necessary
control tests.
(d) Sterility tests are
exclusively based upon the principle that in case the bacteria are
strategically placed in a specific medium that caters for the requisite
nutritive material and water, and maintained duly at a favourable temperature
(37 ± 2°C), the microbes have a
tendency to grow, and their legitimate presence may be clearly indicated by the
appearance of a turbidity in the
originally clear medium.
(e) Extent
of probability in the detection of viable microorganisms for the tests for sterility usually increases
with the actual number supposedly present in a given quantity of the
preparation under examination, and is found to vary according to the species of
microorganisms present. However, extremely low
levels of contamination cannot
be detected conveniently on the basis of random sampling of a batch.*
(f) In
case, observed contamination is not quite uniform throughout the batch, random
sampling cannot detect contamination with absolute certainty. Therefore, compliance with the tests for sterility
individually cannot certify absolute
assurane of freedom from microbial contamination. Nevertheless, greater assurance of sterility should
invariably originate from reliable stringent manufacturing procedures vis-a-vis strict compliance with Good
Manufacturing Practices (GMPs).
(g) Tests for sterility are
adequately designed to reveal the presence of microorganisms in the ‘samples’ used in the tests. However,
the interpretation of results is solely based upon the assumption that the contents of each and every
container in the batch, had they been tested actually, would have complied with the tests. As it is not practically possible to test every
container, a sufficient number of containers must be examined to give a suitable
degree of confidence in the ultimate results obtained of the tests.
(h) It
has been duly observed that there exists no
definite sampling plan for applying the tests to a specified proportion of
discrete units selected carefully from a batch is capable of demonstrating that almost all of the untested units are in fact sterile absolutely.
Therefore, it is indeed quite pertinent that while determining the number of units to be tested, the
manufacturer must have adequate regar to the environment parameters of
manufacture, the volume of preparation per container together with other
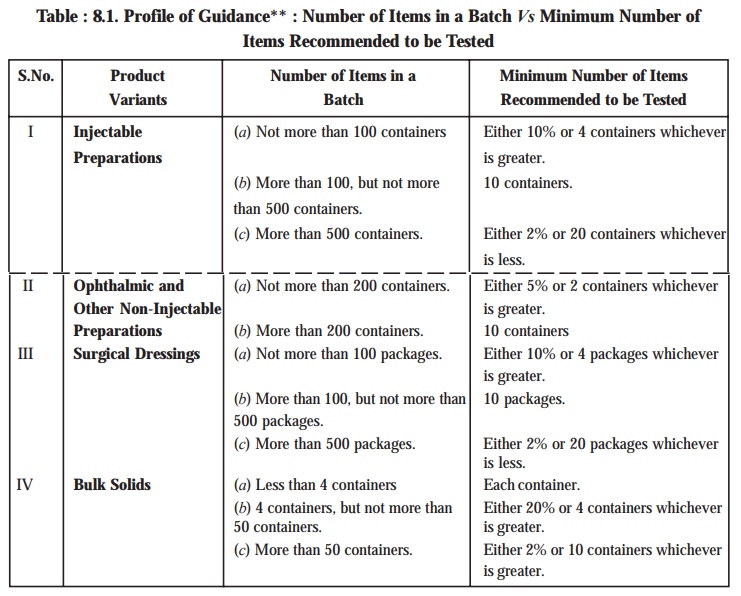
special considerations specific to the preparation under investigation. For
this Table 8.1 records the guidance on the exact number of items recommended to
be tested with regard to the number of items in the batch on the assumption
that the preparation has been duly manufactured under specified stringent
parameters designed meticulously to exclude any untoward contamination.
TEST FOR
STERILITY : PHARMACEUTICAL PRODUCTS
In a
broader perspective the wide-spectrum of the pharmaceutical products, both pure and dosage forms, may be
accomplished by adopting any one of the following two well-recognized, time-tested, and universally accepted methods,
namely :
(a) Membrane
Filtration, and
(b) Direct
Inoculation.
These two methods stated above shall now be
treated individually in the sections that follows :
Related Topics
