Synthesis of Cholesterol
| Home | | Biochemistry |Chapter: Biochemistry : Cholesterol, Lipoprotein, and Steroid Metabolism
Cholesterol is synthesized by virtually all tissues in humans, although liver, intestine, adrenal cortex, and reproductive tissues, including ovaries, testes, and placenta, make the largest contributions to the body’s cholesterol pool.
SYNTHESIS OF CHOLESTEROL
Cholesterol is synthesized by virtually all tissues in humans, although liver, intestine, adrenal cortex, and reproductive tissues, including ovaries, testes, and placenta, make the largest contributions to the body’s cholesterol pool. As with fatty acids, all the carbon atoms in cholesterol are provided by acetyl coenzyme A (CoA), and nicotinamide adenine dinucleotide phosphate (NADPH) provides the reducing equivalents. The pathway is endergonic, being driven by hydrolysis of the high-energy thioester bond of acetyl CoA and the terminal phosphate bond of adenosine triphosphate (ATP). Synthesis requires enzymes in both the cytosol and the membrane of the smooth endoplasmic reticulum (ER). The pathway is responsive to changes in cholesterol concentration, and regulatory mechanisms exist to balance the rate of cholesterol synthesis within the body against the rate of cholesterol excretion. An imbalance in this regulation can lead to an elevation in circulating levels of plasma cholesterol, with the potential for vascular disease.
A. Synthesis of 3-hydroxy-3-methylglutaryl coenzyme A
The first two reactions
in the cholesterol synthetic pathway are similar to those in the pathway that
produces ketone bodies (see Figure 16.22). They result in the production of
3-hydroxy-3-methylglutaryl CoA ([HMG CoA] Figure 18.3). First, two acetyl CoA
molecules condense to form acetoacetyl CoA. Next, a third molecule of acetyl
CoA is added by HMG CoA synthase, producing HMG CoA, a six-carbon compound.
[Note: Liver parenchymal cells contain two isoenzymes of the synthase. The
cytosolic enzyme participates in cholesterol synthesis, whereas the
mitochondrial enzyme functions in the pathway for ketone body synthesis.]
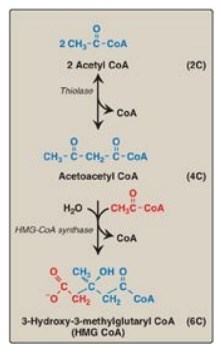
Figure 18.3 Synthesis of HMG CoA. CoA = coenzyme A.
B. Synthesis of mevalonate
The next step, the
reduction of HMG CoA to mevalonate, is catalyzed by HMG CoA reductase and is
the rate-limiting and key regulated step in cholesterol synthesis. It occurs in
the cytosol, uses two molecules of NADPH as the reducing agent, and releases
CoA, making the reaction irreversible (Figure 18.4). [Note: HMG CoA reductase
is an integral membrane protein of the ER, with its catalytic domain projecting
into the cytosol.] Regulation of reductase activity is discussed below.
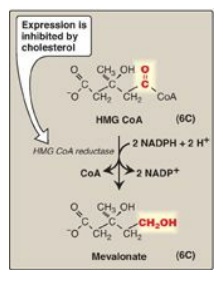
Figure 18.4 Synthesis of mevalonate.
HMG CoA = hydroxymethylglutaryl coenzyme A; NADP(H) = nicotinamide adenine
dinucleotide phosphate.
C. Synthesis of cholesterol
The reactions and
enzymes involved in the synthesis of cholesterol from mevalonate are
illustrated in Figure 18.5. [Note: The numbers shown in brackets below
correspond to numbered reactions shown in this figure.]
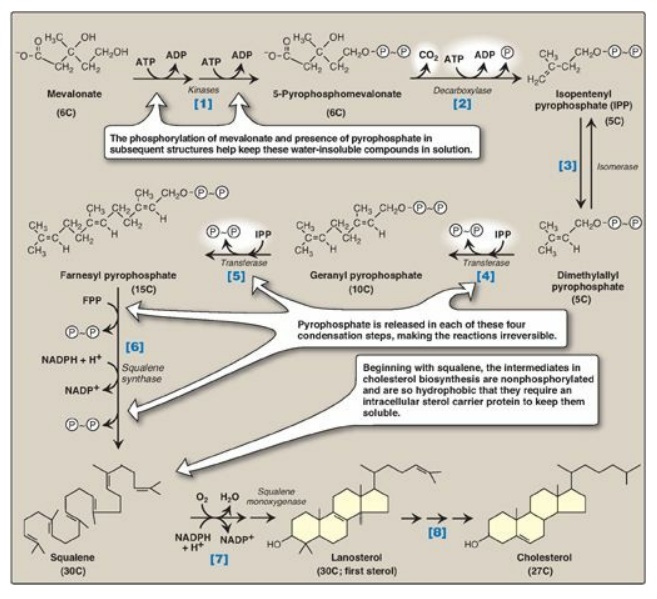
Figure 18.5 Synthesis of cholesterol from mevalonate. ADP = adenosine diphosphate; P = phosphate; P~P = pyrophosphate; NADP(H) = nicotinamide adenine dinucleotide phosphate.
[1] Mevalonate is
converted to 5-pyrophosphomevalonate in two steps, each of which transfers a
phosphate group from ATP.
[2] A five-carbon
isoprene unit, isopentenyl pyrophosphate (IPP), is formed by the
decarboxylation of 5-pyrophosphomevalonate. The reaction requires ATP. [Note:
IPP is the precursor of a family of molecules with diverse functions, the
isoprenoids. Cholesterol is a sterol isoprenoid. Nonsterol isoprenoids include
dolichol and ubiquinone, or coenzyme Q.]
[3] IPP is isomerized
to 3,3-dimethylallyl pyrophosphate (DPP).
[4] IPP and DPP
condense to form ten-carbon geranyl pyrophosphate (GPP).
[5] A second molecule
of IPP then condenses with GPP to form 15-carbon farnesyl pyrophosphate (FPP).
[Note: Covalent attachment of farnesyl to proteins, a process known as
“prenylation,” is one mechanism for anchoring proteins (such as ras) to plasma
membranes.]
[6] Two molecules of
FPP combine, releasing pyrophosphate, and are reduced, forming the 30-carbon
compound squalene. [Note: Squalene is formed from six isoprenoid units. Because
three ATP are hydrolyzed per mevalonate residue converted to IPP, a total of 18
ATP are required to make the polyisoprenoid squalene.]
[7] Squalene is
converted to the sterol lanosterol by a sequence of reactions catalyzed by
ER-associated enzymes that use molecular oxygen and NADPH. The hydroxylation of
linear squalene triggers the cyclization of the structure to lanosterol.
[8] The conversion of
lanosterol to cholesterol is a multistep, ER-associated process involving
shortening of the side-chain, oxidative removal of methyl groups, reduction of
double bonds, and migration of a double bond. Smith-Lemli-Opitz syndrome
(SLOS), an autosomal-recessive disorder of cholesterol biosynthesis, is caused
by a partial deficiency in 7-dehydrocholesterol-7-reductase, the enzyme that
reduces the double bond in 7-dehydrocholesterol (7-DHC), thereby converting it
to cholesterol. SLOS is one of several multisystem, embryonic malformation
syndromes associated with impaired cholesterol synthesis. [Note: 7-DHC is
converted to vitamin D3 in the skin.]
D. Regulation of cholesterol synthesis
HMG CoA reductase is
the major control point for cholesterol biosynthesis and is subject to
different kinds of metabolic control.
1. Sterol-dependent regulation of gene expression: Expression of the gene for HMG CoA
reductase is controlled by the transcription factor, SREBP-2 (sterol regulatory
element–binding protein-2) that binds DNA at the cis-acting sterol regulatory
element (SRE) upstream of the reductase gene. SREBP-2 is an integral protein of
the ER membrane, and associates with a second ER membrane protein, SCAP (SREBP
cleavage–activating protein). When sterol levels in the cell are low, the
SREBP–SCAP complex moves from the ER to the Golgi. In the Golgi membrane,
SREBP-2 is sequentially acted upon by two proteases, which generate a soluble
fragment that enters the nucleus, binds the SRE, and functions as a
transcription factor. This results in increased synthesis of HMG CoA reductase
and, therefore, increased cholesterol synthesis (Figure 18.6). If sterols are
abundant, however, they bind SCAP at its sterol-sensing domain and induce the
binding of SCAP to yet other ER membrane proteins, the insigs (insulin-induced
gene [products]). This results in the retention of the SCAP–SREBP complex in
the ER, thereby preventing the activation of SREBP-2, and leading to
downregulation of cholesterol synthesis. [Note: SREBP-1 upregulates expression
of enzymes involved in fatty acid synthesis in response to insulin.]
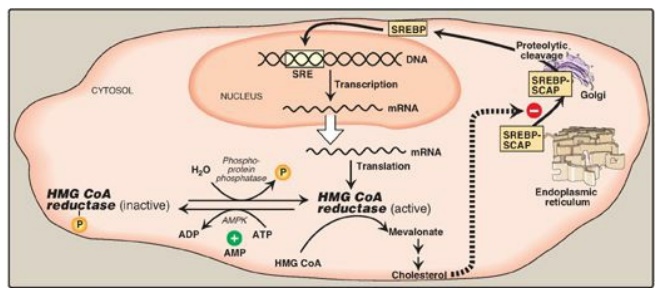
Figure 18.6 Regulation of hydroxymethylglutaryl coenzyme A (HMG CoA) reductase. SRE = sterol regulatory element; SREBP = sterol regulatory element-binding protein; SCAP = SREBP cleavage-activating protein; AMPK = adenosine monophosphate-activated protein kinase; ADP = adenosine diphosphate; P = phosphate; mRNA = messenger RNA.
2. Sterol-accelerated enzyme degradation: The reductase itself is a
sterol-sensing integral protein of the ER membrane. When sterol levels in the
cell are high, the enzyme binds to insig proteins. Binding leads to ubiquitination
and proteasomal degradation of the reductase.
3. Sterol-independent
phosphorylation/dephosphorylation: HMG CoA reductase activity is controlled covalently
through the actions of adenosine monophosphate (AMP)-activated protein kinase
([AMPK]) and a phosphoprotein phosphatase (see Figure 18.6). The phosphorylated
form of the enzyme is inactive, whereas the dephosphorylated form is active.
[Note: Because AMPK is activated by AMP, cholesterol synthesis, like fatty acid
synthesis, is decreased when ATP availability is decreased.]
4. Hormonal regulation: The amount of HMG CoA reductase is controlled hormonally. An increase in insulin and thyroxine favors upregulation of the expression of the gene for the reductase. Glucagon and the glucocorticoids have the opposite effect.
5. Inhibition by drugs: The statin drugs (atorvastatin,
fluvastatin, lovastatin, pravastatin, rosuvastatin, and simvastatin) are
structural analogs of HMG CoA, and are (or are metabolized to) reversible,
competitive inhibitors of HMG CoA reductase (Figure 18.7). They are used to
decrease plasma cholesterol levels in patients with hypercholesterolemia.
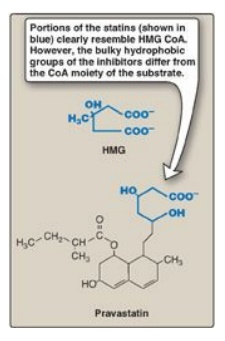
Figure 18.7 Structural
similarity of hydroxymethylglutaric acid (HMG) and pravastatin, a clinically
useful cholesterol-lowering drug of the “statin” family. CoA = coenzyme A.
Related Topics
