Synthesis and Degradation of Glycosphingolipids
| Home | | Biochemistry |Chapter: Biochemistry : Phospholipid, Glycosphingolipid, and Eicosanoid Metabolism
Synthesis of glycosphingolipids occurs primarily in the Golgi by sequential addition of glycosyl monomers transferred from UDP-sugar donors to the acceptor molecule. The mechanism is similar to that used in glycoprotein synthesis.
SYNTHESIS AND DEGRADATION OF GLYCOSPHINGOLIPIDS
Synthesis of
glycosphingolipids occurs primarily in the Golgi by sequential addition of
glycosyl monomers transferred from UDP-sugar donors to the acceptor molecule.
The mechanism is similar to that used in glycoprotein synthesis.
A. Enzymes involved in synthesis
The enzymes involved in
the synthesis of glycosphingolipids are glycosyltransferases that are specific
for the type and location of the glycosidic bond formed. [Note: These enzymes
can recognize both glycosphingolipids and glycoproteins as substrates.]
B. Addition of sulfate groups
A sulfate group from the sulfate carrier 3I -phosphoadenosine-5I -phosphosulfate ([PAPS], Figure 17.16 ) is added by a sulfotransferase to the 3I -hydroxyl group of the galactose in a galactocerebroside, forming the sulfatide galactocerebroside 3-sulfate (Figure 17.17 ). [Note: PAPS is also the sulfur donor in glycosaminoglycan synthesis and steroid hormone catabolism.] An overview of the synthesis of sphingolipids is shown in Figure 17.18.
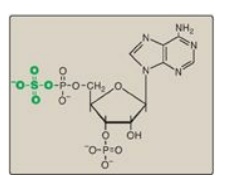
Figure 17.16 Structure of 3I -phosphoadenosine-5I -phosphosulfate.
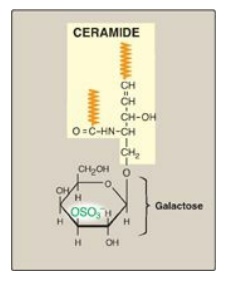
Figure 17.17 Structure of galactocerebroside 3-sulfate. ( is a hydrophobic hydrocarbon chain.)
is a hydrophobic hydrocarbon chain.)
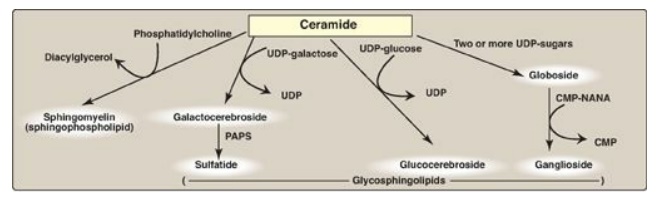
Figure 17.18 Overview of sphingolipid synthesis. UDP = uridine diphosphate; CMP = cytidine monophosphate; NANA = N-acetylneuraminic acid; PAPS = 3I -phosphoadenosine-5 -phosphosulfate.
C. Degradation of glycosphingolipids
Glycosphingolipids are internalized by endocytosis as described for the glycosaminoglycans. All of the enzymes required for the degradative process are present in lysosomes, which fuse with the endocytotic vesicles. The lysosomal enzymes hydrolytically and irreversibly cleave specific bonds in the glycosphingolipid. As seen with the glycosaminoglycans and glycoproteins, degradation is a sequential process following the rule “last on, first off,” in which the last group added during synthesis is the first group removed in degradation. [Note: Defects in the degradation of the polysaccharide chains in these three glycoconjugates, therefore, result in lysosomal storage diseases.]
D. Sphingolipidoses
In a normal individual, synthesis and degradation of glycosphingolipids are balanced, so that the amount of these compounds present in membranes is constant. If a specific lysosomal acid hydrolase required for degradation is partially or totally missing, a sphingolipid accumulates. Lysosomal lipid storage diseases caused by these deficiencies are called sphingolipidoses. The result of a specific acid hydrolase deficiency may be seen dramatically in nerve tissue, where neurologic deterioration can lead to early death. Figure 17.20 provides an outline of the pathway of sphingolipid degradation and descriptions of some sphingolipidoses. [Note: Some sphingolipidoses can also result from defects in lysosomal activator proteins (for example, the saposins) that facilitate access of the hydrolases to short carbohydrate chains as degradation proceeds.]
1. Common properties: A specific lysosomal hydrolytic
enzyme is deficient in the classic form of each disorder. Therefore, usually
only a single sphingolipid (the substrate for the deficient enzyme) accumulates
in the involved organs in each disease. [Note: The rate of biosynthesis of the
accumulating lipid is normal.] The disorders are progressive and, although many
are fatal in childhood, extensive phenotypic variability is seen leading to the
designation of different clinical types, such as Types A and B in Niemann-Pick
disease. Genetic variability is also seen because a given disorder can be
caused by any one of a variety of mutations within a single gene. The
sphingolipidoses are autosomal-recessive diseases, except for Fabry disease,
which is X linked. The incidence of the sphingolipidoses is low in most
populations, except for Gaucher and Tay-Sachs diseases, which, like
Niemann-Pick disease, show a high frequency in the Ashkenazi Jewish population.
[Note: Tay-Sachs also has a high frequency in Irish American, French Canadian,
and Louisiana Cajun populations.]

Figure 17.19 Aspirated bone
marrow cells from a patient with Gaucher disease.
2. Diagnosis and treatment: A specific sphingolipidosis can be
diagnosed by measuring enzyme activity in cultured fibroblasts or peripheral
leukocytes or by analysis of DNA. Histologic examination of the affected tissue
is also useful. [Note: Shell-like inclusion bodies are seen in Tay-Sachs, and a
wrinkled tissue paper appearance of the cytosol is seen in Gaucher disease
(Figure 17.19).] Prenatal diagnosis, using cultured amniocytes or chorionic villi,
is available. Gaucher disease, in which macrophages become engorged with
glucocerebroside, and Fabry disease, in which globosides accumulate in the
vascular endothelial lysosomes of the brain, heart, kidneys, and skin, are
treated by recombinant human enzyme replacement therapy, but the monetary cost
is extremely high. Gaucher has also been treated by bone marrow transplantation
(because macrophages are derived from hematopoietic stem cells) and by
substrate reduction therapy through pharmacologic reduction of
glucosylcer-amide, the substrate for the deficient enzyme.
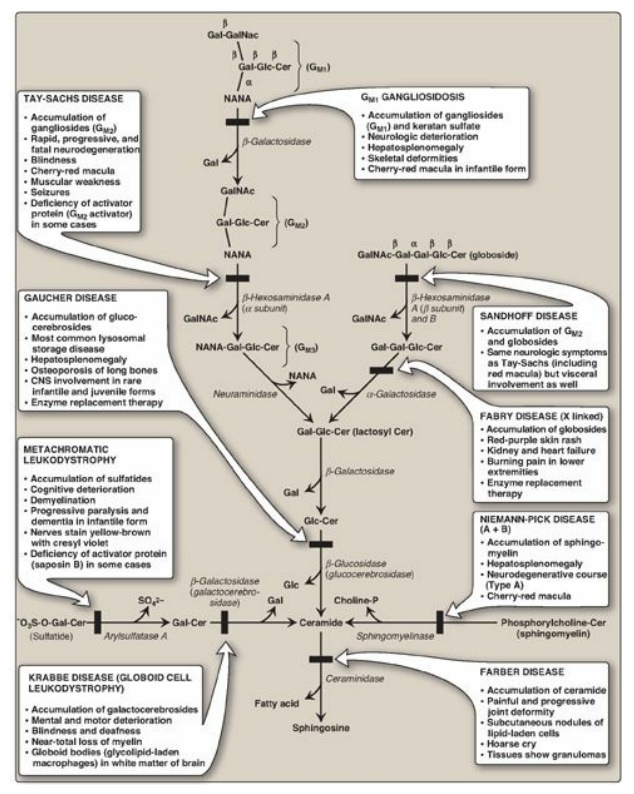
Figure 17.20 Degradation of
sphingolipids showing the lysosomal enzymes affected in related genetic
diseases, the sphingolipidoses. All of the diseases are autosomal recessive
except Fabry disease, which is X linked, and all can be fatal in early life.
Cer = ceramide; Gal = galactose; Glc = glucose; GalNAc = N-acetylgalactosamine;
NANA = N-acetylneuraminic acid; CNS = central nervous system. SO42-
= sulfate.
Related Topics
