Co-and Posttranslational Modification of Polypeptide Chains
| Home | | Biochemistry |Chapter: Biochemistry : Protein Synthesis
Many polypeptide chains are covalently modified, either while they are still attached to the ribosome (cotranslational) or after their synthesis has been completed (posttranslational).
CO-AND POSTTRANSLATIONAL MODIFICATION OF
POLYPEPTIDE CHAINS
Many polypeptide chains
are covalently modified, either while they are still attached to the ribosome
(cotranslational) or after their synthesis has been completed
(posttranslational). These modifications may include removal of part of the
translated sequence or the covalent addition of one or more chemical groups
required for protein activity. Examples of such modifications are listed below.
A. Trimming
Many proteins destined
for secretion from the cell are initially made as large, precursor molecules
that are not functionally active. Portions of the protein chain must be removed
by specialized endoproteases, resulting in the release of an active molecule.
The cellular site of the cleavage reaction depends on the protein to be
modified. Some precursor proteins are cleaved in the endoplasmic reticulum or
the Golgi apparatus; others are cleaved in developing secretory vesicles (for
example, insulin; see Figure 23.4); and still others, such as collagen, are
cleaved after secretion.
B. Covalent attachments
Proteins may be
activated or inactivated by the covalent attachment of a variety of chemical
groups (Figure 31.16). Examples include the following.
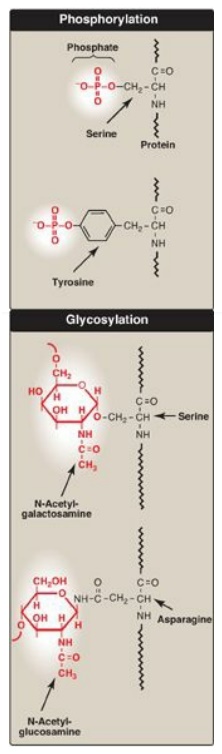
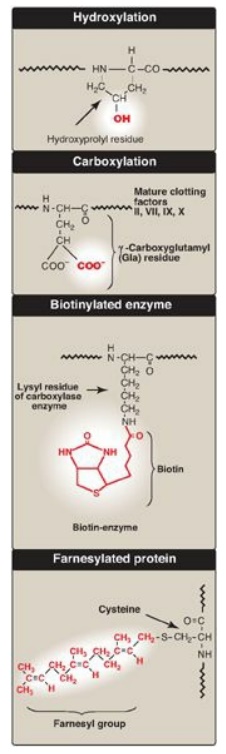
Figure 31.16 Covalent
modifications of some amino acid residues.
1. Phosphorylation: Phosphorylation occurs on the
hydroxyl groups of serine; threonine; or, less frequently, tyrosine residues in
a protein. This phosphorylation is catalyzed by one of a family of protein
kinases and may be reversed by the action of cellular protein phosphatases. The
phosphorylation may increase or decrease the functional activity of the
protein. Several examples of phosphorylation reactions have been previously
discussed (for example, see Chapter 11, for the regulation of glycogen
synthesis and degradation).
2. Glycosylation: Many of the proteins that are destined to become
part of a plasma membrane or to be secreted from a cell have carbohydrate
chains added en bloc to the amide nitrogen of asparagine (N-linked) or built
sequentially on the hydroxyl groups of serine, threonine, or hydroxylysine
(O-linked). N-glycosylation occurs in the endoplasmic reticulum and
O-glycosyation in the Golgi. (The process of producing such glycoproteins.)
Glycosylation is also used to target proteins to the matrix of lysosomes.
Lysosomal acid hydrolases are modified by the phosphorylation of mannose
residues at carbon 6.
3. Hydroxylation: Proline and lysine residues of the a chains of
collagen are extensively hydroxylated by vitamin C–dependent hydroxylases in
the endoplasmic reticulum.
4. Other covalent modifications: These may be required for the
functional activity of a protein. For example, additional carboxyl groups can
be added to glutamate residues by vitamin K–dependent carboxylation. The
resulting g-carboxyglutamate (Gla) residues are essential for the activity of
several of the blood-clotting proteins. (See Premium Chapter 34.) Biotin is
covalently bound to the e-amino groups of lysine residues of biotin-dependent
enzymes that catalyze carboxylation reactions such as pyruvate carboxylase.
Attachment of lipids, such as farnesyl groups, can help anchor proteins to
membranes. Many eukaryotic proteins are cotranslationally acetylated at the
N-end. [Note: Reversible acetylation of histone proteins influences gene
expression.]
C. Protein folding
Proteins must fold to
assume their functional, native state. Folding can be spontaneous (as a result
of the primary structure) or facilitated by proteins known as chaperones.
D. Protein degradation
Proteins that are
defective (for example, misfolded) or destined for rapid turnover are often
marked for destruction by ubiquitination, the attachment of chains of a small,
highly conserved protein, called ubiquitin (see Figure 19.3). Proteins marked
in this way are rapidly degraded by a cellular component known as the
proteasome, which is a macromolecular, ATP-dependent, proteolytic system
located in the cytosol. [Note: The DF508 mutation seen in CF causes misfolding
of the CFTR protein, resulting in its proteasomal degradation.]
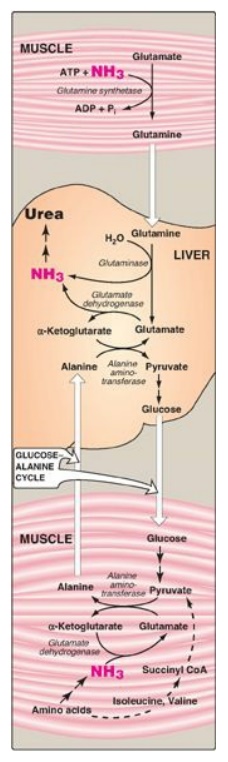
Figure 19.13 Transport of ammonia (NH3) from muscle to the liver. ADP = adenosine diphosphate; Pi = inorganic phosphate; CoA = coenzyme A.
Related Topics
