Design of Dosage Regimens
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Bioavailability and Bioequivalence
Dosage regimen is defined as the manner in which a drug is taken. For some drugs like analgesics, hypnotics, antiemetics, etc., a single dose may provide effective treatment.
DESIGN OF DOSAGE REGIMENS
Dosage regimen is defined as the manner in which
a drug is taken. For some drugs like analgesics,
hypnotics, antiemetics, etc., a single dose may provide effective treatment.
However, the duration of most illnesses is longer than the therapeutic effect
produced by a single dose. In such cases, drugs are required to be taken on a
repetitive basis over a period of time depending upon the nature of illness.
Thus, for successful therapy, design of an optimal multiple dosage regimen is
necessary. An optimal multiple dosage regimen
is the one in which the drug is administered in suitable doses (by a suitable
route), with sufficient frequency that ensures maintenance of plasma
concentration within the therapeutic window (without excessive fluctuations and
drug accumulation) for the entire duration of therapy. For some drugs like antibiotics, a minimum effective concentration
should be maintained at all times and for drugs with narrow therapeutic indices
like phenytoin, attempt should be made not to exceed toxic concentration.
Approaches to Design of Dosage Regimen
The various approaches employed in designing a
dosage regimen are –
1. Empirical Dosage Regimen – is designed by the physician based on empirical clinical data,
personal experience and clinical observations. This approach is, however, not
very accurate.
2. Individualized Dosage Regimen – is the most accurate approach and is based on the pharmacokinetics
of drug in the individual patient. The approach is suitable for hospitalised
patients but is quite expensive.
3. Dosage Regimen on Population
Averages – This is the most often used
approach. The method is based on one of the two models –
(a) Fixed model – here, population average pharmacokinetic parameters are used directly
to calculate the dosage regimen.
(b) Adaptive model – is based on both population average pharmacokinetic parameters
of the drug as well as patient variables such as weight, age, sex, body surface
area and known patient pathophysiology such as renal disease.
In designing a dosage regimen based on population
averages —
1. It is assumed that all pharmacokinetic
parameters of the drug remain constant during the course of therapy once a
dosage regimen is established. The same becomes invalid if any change is
observed.
2. The calculations are based on open
one-compartment model which can also be applied to two compartment model if β is used instead of KE, and Vd,ss instead of Vd
while calculating the regimen.
Irrespective of the route of administration and
complexity of pharmacokinetic equations, the two major parameters that can be
adjusted in developing a dosage regimen are —
1. The dose size — the quantity of drug administered each time,
and
2. The dosing frequency — the time interval between doses.
Both parameters govern the amount of drug in the
body at any given time.
Dose Size
The magnitude of both therapeutic and toxic
responses depends upon dose size. Dose size calculation also requires the
knowledge of amount of drug absorbed after administration of each dose. Greater
the dose size, greater the fluctuations between Css,max and Css,min
during each dosing interval and greater the chances of toxicity (Fig. 12.1).
For drugs administered chronically, dose size calculation is based on average
steady state blood levels and is computed from equation 12.8.
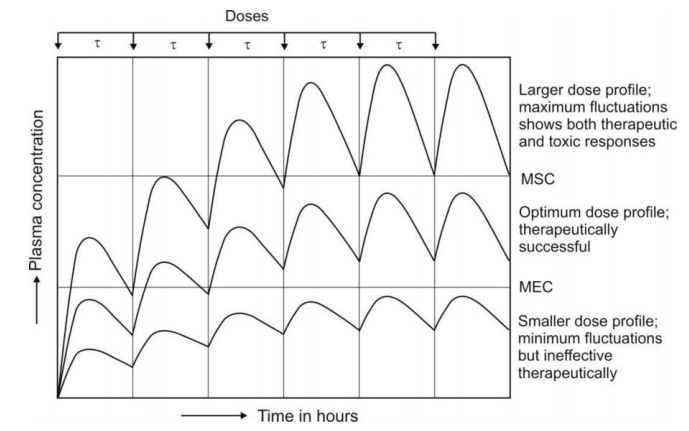
Fig. 12.1 Schematic representation of influence of dose size on plasma
concentration-time profile after oral administration of a drug at fixed
intervals of time.
Dosing Frequency
The dose
interval (inverse of dosing frequency)
is calculated on the basis of half-life of the drug. If the interval is
increased and the dose is unchanged, Cmax, Cmin and Cav
decrease but the ratio Cmax/Cmin increases. Opposite is
observed when dosing interval is reduced or dosing frequency increased. It also
results in greater drug accumulation in the body and toxicity (Fig. 12.2).
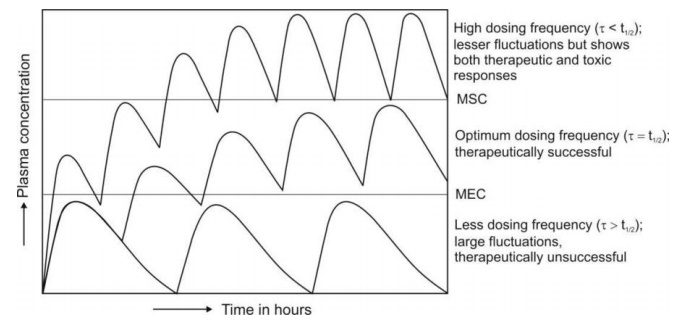
Fig. 12.2 Schematic representation of the
influence of dosing frequency on plasma concentration-time
profile obtained after oral administration of fixed doses of a drug.
A proper balance between both dose size and dosing
frequency is often desired to attain steady-state concentration with minimum
fluctuations and to ensure therapeutic efficacy and safety. The same cannot be
obtained by giving larger doses less frequently. However, administering smaller
doses more frequently results in smaller fluctuations.
Generally speaking, every subsequent dose should be
administered at an interval equal to half-life of the drug. A rule of thumb is
that –
·
For drugs with wide therapeutic index such as
penicillin, larger doses may be
administered at relatively longer
intervals (more than the half-life of drug) without any toxicity problem
·
For drugs with narrow therapeutic index such as
digoxin, small doses at frequent intervals (usually less than the half-life of the drug) is better
to obtain a profile with least
fluctuations which is similar to that observed with constant rate infusion or
controlled-release system.
Drug Accumulation During Multiple Dosing
Consider the amount of drug in the body-time
profile shown in Fig. 12.3. obtained after i.v. multiple dosing with dosing
interval equal to one t½.
After the administration of first dose Xo
at τ = 0, the amount of drug in the body will be X = 1Xo. At the
next dosing interval when X = ½Xo, the amount of drug remaining in
the body, administration of the next i.v. dose raises the body content to X = Xo
+ ½Xo i.e. drug accumulation occurs in the body. Thus, accumulation occurs because drug from
previous doses has not been removed
completely. As the amount of drug in the body rises gradually due to accumulation, the rate of
elimination also rises proportionally until a steady-state or plateau is
reached when the rate of drug entry into the body equals the rate of exit. The
maximum and minimum values of X i.e. Xss,max and Xss,min
approach respective asymptotes at plateau. It is interesting to note that at
plateau, Xss,min = 1Xo and Xss,max = 2Xo
i.e. Xss,min equals the amount of drug in the body after the first
dose and Xss,max equals twice the first dose. Also (Xss,max
– Xss,min) = Xo and Xss,max/Xss,min
= 2. All this applies only when τ = t½ and drug is administered
intravenously. When τ < t½, the degree
of accumulation is greater and vice-versa.
Thus, the extent
to which a drug accumulates in the body during multiple dosing is
independent of dose size, and is a
function of –
·
Dosing interval, and
·
Elimination half-life.
The extent to which a drug will accumulate with any dosing interval in a patient can be derived from information obtained with a single dose and is given by accumulation index Rac as:
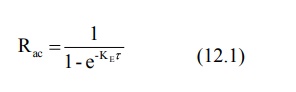
Fig.12.3 Accumulation of drug in the body during multiple dose regimen of i.v.
bolus with dosing interval equal to
one half-life of the drug. Approximately 5 half-lives are required for
attainment of steady-state.
Time to Reach Steady-State During Multiple Dosing
The time required to reach steady-state depends
primarily upon the half-life of the drug. Provided Ka >> KE,
the plateau is reached in approximately 5
half-lives. This is called as plateau
principle. It also means that the rate at which the multiple dose
steady-state is reached is determined
only by KE. The time taken to reach steady-state is independent of
dose size, dosing interval and number of doses.
Maximum and Minimum Concentration During Multiple Dosing
If n is the number of doses administered, the Cmax
and Cmin obtained on multiple dosing after the nth dose is given as:
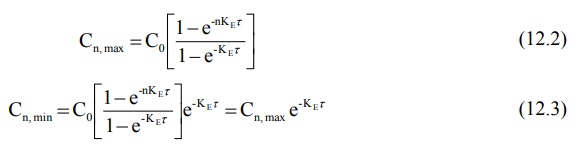
The maximum and minimum concentration of drug in
plasma at steady-state are found by following equations:
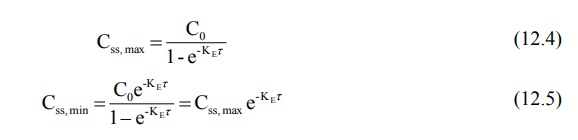
where Co = concentration that would be attained
from instantaneous absorption and distribution (obtained by extrapolation of
elimination curve to time zero). Equations 12.2 to 12.5 can also be written in
terms of amount of drug in the body. Fraction of dose absorbed, F, should be
taken into account in such equations.
Fluctuation is defined as the ratio Cmax/Cmin. Greater the ratio, greater the fluctuation. Like accumulation, it
depends upon dosing frequency and half-life of the drug. It also depends upon
the rate of absorption. The greatest fluctuation is observed when the drug is
given as i.v. bolus. Fluctuations are small when the drug is given
extravascularly because of continuous absorption.
Average Concentration and Body Content on Multiple Dosing to Steady-State
The average drug concentration at steady-state Css,av is a
function of –
·
The maintenance dose Xo,
·
The fraction of dose absorbed F,
·
The dosing interval τ and
·
Clearance ClT (or Vd
and KE or t½) of the drug.
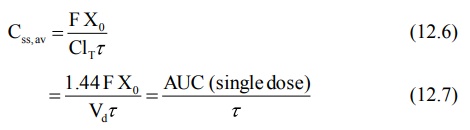
where the coefficient 1.44 is the reciprocal of
0.693 in equation 12.7. AUC is the area under the curve following a single
maintenance dose. Equation 12.7 can be used to calculate maintenance dose of a
drug to achieve a desired concentration. Since X = Vd C, the body
content at steady-state is given as:
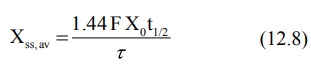
These average values are not arithmetic mean of Css,max
and Css,min since the plasma drug concentration declines
exponentially.
Loading and Maintenance Doses
A drug does not show therapeutic activity unless it
reaches the desired steady-state. It takes about 5 half-lives to attain it and
therefore the time taken will be too long if the drug has a long half-life.
Plateau can be reached immediately by administering a dose that gives the
desired steady-state instantaneously before the commencement of maintenance
doses Xo.
Such an initial or first dose intended to be therapeutic is called as priming dose or loading dose Xo,L. A simple equation
for calculating loading dose is:
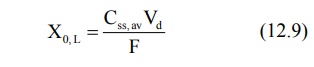
After e.v. administration, Cmax is
always smaller than that after i.v. administration and hence loading dose is
proportionally smaller. For drugs having low therapeutic indices, the loading
dose may be divided into smaller doses to be given at various intervals before
the first maintenance dose. When Vd is not known, loading dose may
be calculated by the following equation:
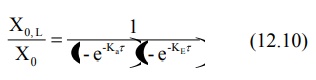
The above equation applies when Ka
>> KE and drug is distributed rapidly. When the drug is given
i.v. or when absorption is extremely rapid, the absorption phase is neglected
and the above equation reduces to accumulation
index:
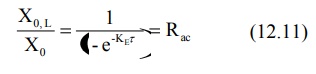
The ratio of loading dose to maintenance dose Xo,L/Xo is
called as dose ratio. As a rule, when τ = t½,
dose ratio should be equal to 2.0 but must be smaller than 2.0 when τ > t½ and greater
when τ < t½. Fig. 12.4. shows that if loading dose is not
optimum, either too low or too high, the steady-state is attained within
approximately 5 half-lives in a manner similar to when no loading dose is
given.
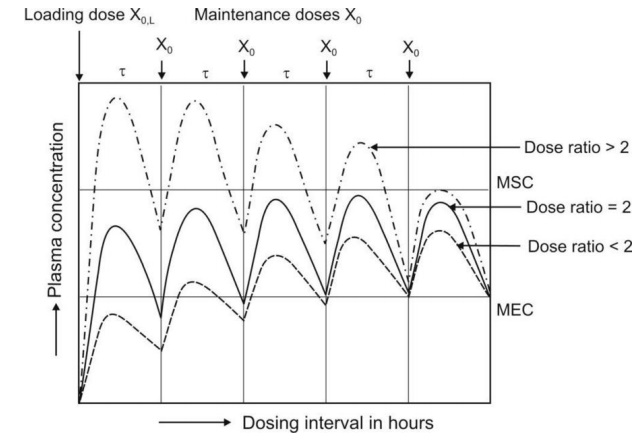
Fig. 12.4 Schematic representation of plasma concentration-time profiles that
result when dose ratio is greater
than 2.0, equal to 2.0 and smaller than 2.0.
The foregoing discussion and the equations expressed
until now apply only to drugs that follow one-compartment kinetics with
first-order disposition. Equations will be complex for multicompartment models.
Maintenance of Drug within the Therapeutic Range
The ease or difficulty in maintaining drug concentration
within the therapeutic window depends upon —
1. The therapeutic index of the
drug
2. The half-life of the drug
3. Convenience of dosing.
It is extremely difficult to maintain such a level
for a drug with short half-life (less than 3 hours) and narrow therapeutic
index e.g. heparin, since the dosing frequency has to be essentially less than
t½. However, drugs such as penicillin (t½ = 0.9 hours)
with high therapeutic index may be given less frequently (every 4 to 6 hours)
but the maintenance dose has to be larger so that the plasma concentration
persists above the minimum inhibitory level.
A drug with intermediate t½ (3 to 8
hours) may be given at intervals τ ≤ t½ if therapeutic index is low and those with high indices
can be given at intervals between 1 to 3 half-lives. Drugs with half-lives
greater than 8 hours are more convenient to dose. Such drugs are usually
administered once every half-life. Steady-state in such cases can be attained
rapidly by administering a loading dose. For drugs with very long half-lives
(above 24 hours) e.g. amlodipine, once daily dose is very convenient.
Design of Dosage Regimen from Plasma Concentrations
If the therapeutic range, apparent Vd
and clearance or half-life of a drug is known, then dosage regimen can be designed
to maintain drug concentration within the specified therapeutic range. The
latter is defined by lower limit (Clower) and an upper limit (Cupper).
The maximum dosing interval, which ideally depends upon the therapeutic index
(can be defined as a ratio of Cupper to Clower) and
elimination half-life of the drug, can be expressed by equation 12.12, a
modification of equation 12.5.
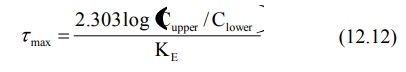
Since KE = 0.693/t½, the above equation can also be written as:
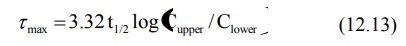
Understandably, the dosing interval selected is
always smaller than τmax. The maximum maintenance dose Xo,max that can be given every τmax is expressed as:

After a convenient dosing interval τ, smaller than τmax has been selected, the maintenance dose is given as:
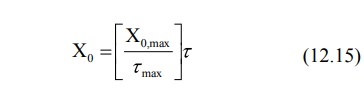
Administration of Xo every τ produces an average steady-state concentration defined by equation
12.7. The value of Css,av can also be calculated from the therapeutic range
according to equation 12.16.

Related Topics
