Bioavailability Enhancement Through Enhancement of Drug Solubility or Dissolution Rate
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Bioavailability and Bioequivalence
Bioavailability Enhancement Through Enhancement of Drug Solubility or Dissolution Rate
BIOAVAILABILITY ENHANCEMENT THROUGH ENHANCEMENT OF DRUG SOLUBILITY OR
DISSOLUTION RATE
There are several ways by which drug solubility or
the dissolution rate can be enhanced.
Some of the widely used methods are discussed
briefly.
1. Micronization: The process involves reducing the
size of the solid drug particles to
1 to 10 microns commonly by spray drying or by use of air attrition methods
(fluid energy or jet mill). The process is also called as micro-milling. Examples
of drugs whose bioavailability have been increased by micronization include
griseofulvin and several steroidal and sulpha drugs.
2. Nanonisation: It‘s a process whereby the drug
powder is converted to nanocrystals
of sizes 200 - 600 nm, e.g. amphotericin B. The main production technologies
currently in use to produce drug nanocrystals yield as a product a dispersion
of drug nanocrystals in a liquid, typically water (called nanosuspension). There
are three basic technologies currently in use to prepare nanoparticles:
i. Pearl milling
ii. Homogenisation in water (wet
milling as in a colloid mill)
iii. Homogenisation in non-aqueous
media or in water with water-miscible liquids.
3. Supercritical Fluid Recrystallization: Another novel nanosizing and solubilisation
technology whose application has increased in recent years is particle size
reduction via supercritical fluid
(SCF) processes. Supercritical fluids (e.g. carbon dioxide) are fluids whose
temperature and pressure are greater than its critical temperature (Tc) and
critical pressure (Tp), allowing it to assume the properties of both a liquid
and a gas. At near-critical temperatures, SCFs are high compressible, allowing
moderate changes in pressure to greatly alter the density and mass transport
characteristics of a fluid that largely determine its solvent power. Once the
drug particles are solubilised within SCF, they may be recrystallised at
greatly reduced particle sizes.
4. Use of Surfactants: Surfactants
are very useful as absorption enhancers and
enhance both dissolution rate as well as permeability of drug. They enhance
dissolution rate primarily by promoting wetting and penetration of dissolution
fluid into the solid drug particles. They are generally used in concentration
below their critical micelle concentration (CMC) values since above CMC, the
drug entrapped in the micelle structure fails to partition in the dissolution
fluid. Nonionic surfactants like polysorbates are widely used. Examples of
drugs whose bioavailability have been increased by use of surfactants in the
formulation include steroids like spironolactone.
5. Use of Salt Forms: Salts
have improved solubility and dissolution characteristics in comparison to the original drug. It is generally accepted that
a minimum difference of 3 units between the pKa value of the group and that of
its counterion is required to form stable
salts. Alkali metal salts of acidic drugs like penicillins and strong acid
salts of basic drugs like atropine are more water-soluble than the parent drug.
Factors that influence salt selection are physical and chemical properties of
the salt, safety of counterion, therapeutic indications and route of
administration.
Salt formation does have its limitations –
·
It is not feasible to form salts
of neutral compounds.
·
It may be difficult to form salts
of very weak bases or acids.
·
The salt may be hygroscopic,
exhibit polymorphism or has poor processing characteristics.
·
Conversion of salt to free acid
or base form of the drug on surface of solid dosage form that prevents or
retards drug release.
·
Precipitation of unionised drug
in the GI milieu that has poor solubility.
6. Use of Precipitation Inhibitors: A
significant increase in free drug concentration above equilibrium solubility results in supersaturation, which can
lead to drug precipitation or crystallization. This can be prevented by use of
inert polymers such HPMC, PVP, PVA, PEG, etc. which act by one or more of the
following mechanisms -
·
Increase the viscosity of
crystallization medium thereby reducing the crystallization rate of drugs.
·
Provide a steric barrier to drug
molecules and inhibit crystallization through specific intermolecular interactions
on growing crystal surfaces.
·
Adsorb onto faces of host
crystals, reduce the crystal growth rate of the host and produce smaller
crystals.
7. Alteration of pH of the Drug Microenvironment: This can be achieved in two ways—in situ salt formation, and addition of
buffers to the formulation e.g. buffered aspirin tablets.
8. Use of Amorphs, Anhydrates, Solvates and Metastable Polymorphs: Depending upon the internal structure of the solid drug, selection of
proper form of drug with greater solubility is important. In general, amorphs
are more soluble than metastable polymorphs, anhydrates are more soluble than
hydrates and solvates are more soluble than non-solvates.
9. Solvent Deposition: In this
method, the poorly aqueous soluble drug such as nifedipine is dissolved in an organic solvent like alcohol and
deposited on an inert, hydrophilic, solid matrix such as starch or
microcrystalline cellulose by evaporation of solvent.
10. Precipitation: In this method, the poorly
aqueous soluble drug such as cyclosporine
is dissolved in a suitable organic solvent followed by its rapid mixing with a
non-solvent to effect precipitation of drug in nanosize particles. The product
so prepared is also called as hydrosol.
11. Selective Adsorption on Insoluble Carriers: A highly active adsorbent such as
the inorganic clays like bentonite can enhance the dissolution rate of
poorly water-soluble drugs such as griseofulvin, indomethacin and prednisone by
maintaining the concentration gradient at its maximum. The two reasons suggested
for the rapid release of drugs from the surface of clays are—the weak physical
bonding between the adsorbate and the adsorbent, and hydration and swelling of
the clay in the aqueous media.
12. Solid Solutions: The three
means by which the particle size of a drug can be reduced to submicron level are—
·
Use of solid solutions,
·
Use of eutectic mixtures, and
·
Use of solid dispersions.
In all these cases, the solute is frequently a
poorly water-soluble drug acting as the guest
and the solvent is a highly water-soluble compound or polymer acting as a host or carrier.
A solid
solution is a binary system
comprising of a solid solute molecularly dispersed in a solid solvent. Since the two components crystallize together
in a homogeneous one phase system,
solid solutions are also called as molecular
dispersions or mixed crystals.
Because of reduction in particle size to the molecular level, solid solutions
show greater aqueous solubility and faster dissolution than eutectics and solid
dispersions. They are generally prepared by fusion method whereby a physical
mixture of solute and solvent are melted together followed by rapid
solidification. Such systems, prepared by
fusion, are often called as melts e.g. griseofulvin-succinic acid (Fig. 11.5). The griseofulvin from such solid solution dissolves 6 to
7 times faster than pure griseofulvin.
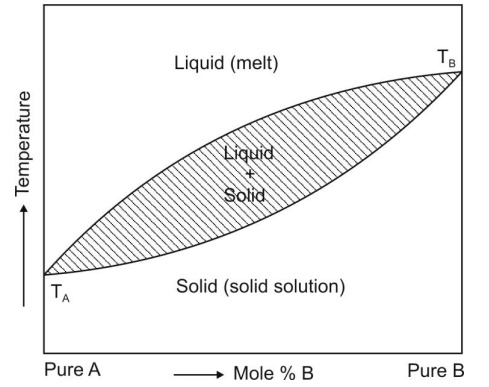
Fig. 11.5 Binary phase diagram for continuous solid solution of A and B. TA and
TB are melting points of pure A and pure B respectively.
If the diameter of solute molecules is less than
60% of diameter of solvent molecules or its volume less than 20% of volume of
solvent molecule, the solute molecule can be accommodated within the
intermolecular spaces of solvent molecules e.g. digitoxin-PEG 6000 solid solution.
Such systems show faster dissolution. When
the resultant solid solution is a
homogeneous transparent and brittle system, it is called as glass solution. Carriers that form glassy structure are citric acid, urea, PVP and
PEG and sugars such as dextrose, sucrose and galactose.
Solid solutions can be classified on two basis –
A. Miscibility between the drug and the carrier – on this basis the solid
solutions are divided into two
categories –
1. Continuous solid solution is the one in which both the drug
and the carrier are miscible in all
proportions. Such a solid solution is not reported in pharmaceutical literature.
2. Discontinuous solid solution is the one where solubility of
each of the component in the other
is limited (see fig. 11.5).
B. Distribution of drug in carrier structure – on this basis the solid
solutions are divided into two
categories –
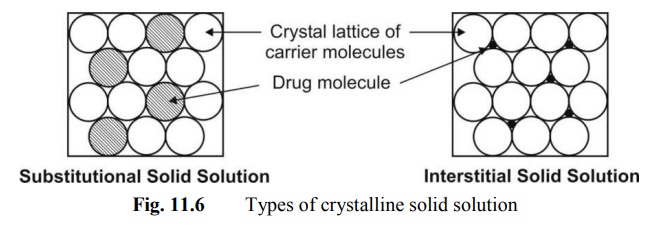
1. Substitutional crystalline
solid solution is the one in which the drug molecules substitute for the carrier molecules in its crystal lattice. This happens when the drug and
carrier molecules are almost of same size.
2. Interstitial crystalline solid
solution is the one in which the drug molecules occupy the interstitial spaces in the crystal lattice of carrier
molecules. This happens
when the size of drug molecule is 40% or less than the size of carrier
molecules (fig. 11.6).
The two mechanisms suggested for enhanced
solubility and rapid dissolution of molecular dispersions are:
·
When the binary mixture is
exposed to water, the soluble carrier dissolves rapidly leaving the insoluble
drug in a state of microcrystalline dispersion of very fine particles, and
·
When the solid solution, which is
said to be in a state of randomly arranged solute and solvent molecules in the
crystal lattice, is exposed to the dissolution fluid, the soluble carrier
dissolves rapidly leaving the insoluble drug stranded at almost molecular
level.
Fig. 11.7 shows a comparison between the
dissolution rates of different forms of griseofulvin.
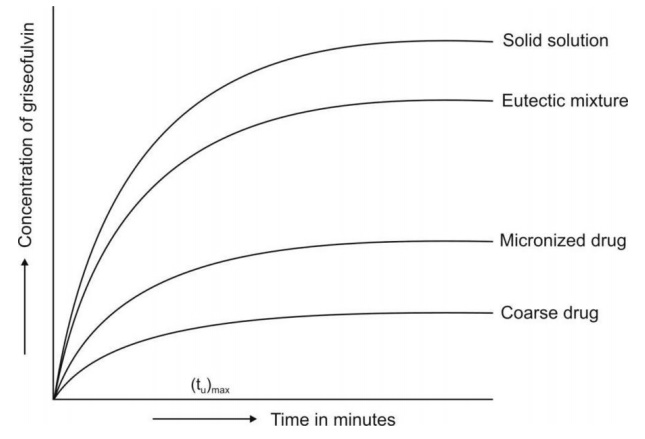
Fig. 11.7 Dissolution rates of griseofulvin as coarse particles, as micronized particles
and as eutectic and solid solution with succinic acid.
13. Eutectic Mixtures: These
systems are also prepared by fusion method. Eutectic melts differ from solid solutions in that the fused melt of
solute-solvent show complete miscibility but negligible solid-solid solubility
i.e. such systems are basically
intimately blended physical mixture
of two crystalline components. A phase diagram of two-component system is
shown in Fig. 11.8. When the eutectic mixture is exposed to water, the soluble
carrier dissolves leaving the drug in a microcrystalline state which
solubilises rapidly.
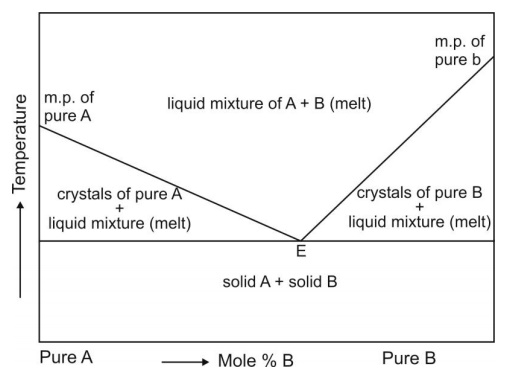
Fig. 11.8 Simple binary phase diagram
showing eutectic point E. The eutectic composition
at point E of substances A and B represents the one having lowest melting
point.
Examples of eutectics include paracetamol-urea,
griseofulvin-urea, griseofulvin-succinic acid, etc. Solid solutions and
eutectics, which are basically melts, are easy to prepare and economical with
no solvents involved. The method, however, cannot be applied to:
·
Drugs which fail to crystallize
from the mixed melt.
·
Drugs which are thermolabile.
·
Carriers such as succinic acid
that decompose at their melting point. The eutectic product is often tacky,
intractable or irregular crystal.
14. Solid Dispersions: These are
generally prepared by solvent or co-precipitation method whereby both
the guest solute and the solid carrier solvent are dissolved in a common
volatile liquid solvent such as alcohol. The liquid solvent is removed by
evaporation under reduced pressure or by freeze-drying which results in
amorphous precipitation of guest in a crystalline carrier. Thus, the basic difference between solid dispersions and solid solutions/eutectics is
that the drug is precipitated out in an amorphous form in the former as opposed
to crystalline form in the latter; e.g.
amorphous sulphathiazole in crystalline urea. Such dispersions are often
called as co-evaporates or co-precipitates. The method is
suitable for thermolabile substances but has a number of disadvantages like
higher cost of processing, use of large quantities of solvent, difficulty in
complete removal of solvent, etc. The carriers used are same as for eutectics
or solid solutions. With glassy materials, the dispersions formed are called as
glass dispersions or glass suspensions. Fig. 11.9 shows
comparative dissolution rates of griseofulvin
from PVP dispersions. Other polymers such as PEG and HPMC are also employed to
prepare solid dispersions of poorly water-soluble drugs such as nifedipine and
itraconazole.
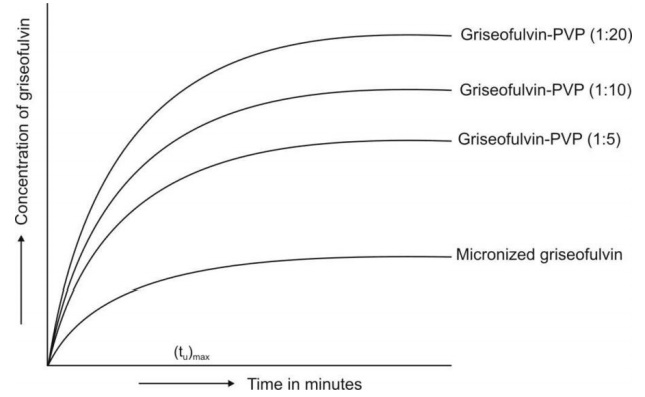
Fig. 11.9 Dissolution rate enhancement of
griseofulvin by solid dispersion technique.
Preparation of solid dispersions also presents
several limitations –
·
Since the carrier is hydrophilic
and the drug is hydrophobic, it is difficult to find a common solvent to
dissolve both components.
·
The product is often soft, waxy
and possesses poor compressibility and flowability.
·
Physical instability of the solid
dispersion.
·
Difficulty in preparation of a
reproducible product.
15. Molecular Encapsulation with Cyclodextrins: The beta- and gamma-cyclodextrins and several of their derivatives are
unique in having the ability to form molecular inclusion complexes with
hydrophobic drugs having poor aqueous solubility. These bucket-shaped
oligosaccharides produced from starch are versatile in having a hydrophobic
cavity of size suitable enough to accommodate the lipophilic drugs as guests;
the outside of the host molecule is relatively hydrophilic (Fig. 11.10). Thus,
the molecularly encapsulated drug has greatly improved aqueous solubility and
dissolution rate. There are several examples of drugs with improved
bioavailability due to such a phenomenon — thiazide diuretics, barbiturates,
benzodiazepines and a number of NSAIDs.
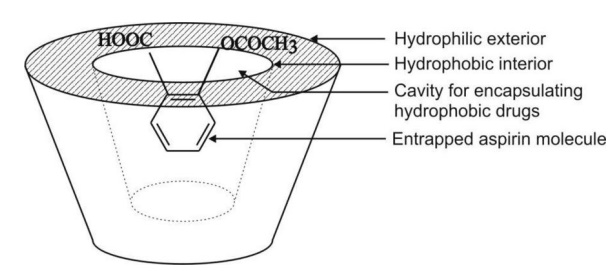
Fig. 11.10 Functional and structural feature
of a cyclodextrin molecule showing an encapsulated
drug.
16. Spherical Crystallization: add some text
Related Topics
