Considerations In In-Vivo Bioavailability Study Design
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Bioavailability and Bioequivalence
When the systemic availability of a drug administered orally is determined in comparison to its intravenous administration, it is called as absolute bioavailability.
CONSIDERATIONS IN IN-VIVO
BIOAVAILABILITY STUDY DESIGN
Bioavailability-Absolute versus Relative
When the systemic availability of a drug
administered orally is determined in comparison to its intravenous administration,
it is called as absolute bioavailability.
It is denoted by symbol F. Its determination is used to characterize a drug‘s
inherent absorption properties from the e.v. site. Intravenous dose is selected
as a standard because the drug is administered directly into the systemic
circulation (100% bioavailability) and avoids absorption step. Intramuscular
dose can also be taken as a standard if the drug is poorly water-soluble. An
oral solution as reference standard has also been used in certain cases.
However, there are several drawbacks of
using oral solution as a standard instead of an i.v. dose —
1. Limits the pharmacokinetic
treatment to one-compartment model only; one cannot apply the most applicable
two-compartment kinetics to the data and all pharmacokinetic parameters cannot
be assessed.
2. Differentiation between the
fraction of dose unabsorbed and that metabolised is difficult.
3. If the rate of oral absorption
is not sufficiently greater than the rate of elimination, the true elimination
rate constant cannot be computed.
At best, when oral solution is used in conjunction
with i.v. route, one can distinguish the dissolution rate limitation in drug
absorption from solid dosage forms.
When the systemic availability of a drug after oral administration is
compared with that of an oral standard of the same drug (such as an aqueous or non-aqueous solution or a suspension), it is
referred to as relative or comparative bioavailability. It is
denoted by symbol Fr. In contrast to absolute bioavailability, it is
used to characterize absorption of a drug from its formulation. F and Fr
are generally expressed in percentages.
Single Dose versus Multiple Dose Studies
The dose to be administered for a bioavailability
study is determined from preliminary clinical experiments. Single dose bioavailability studies are very common. They are easy,
offer less exposure to drugs and are less tedious. However, it is difficult to
predict the steady-state characteristics of a drug and inter-subject
variability with such studies. Moreover, sufficiently long sampling periods are
necessary in order to get reliable estimates of terminal half-life, which is
needed for correct calculation of the total AUC. The better alternative is
thus, multiple dose study which
offers several advantages —
1.
More accurately reflects the
manner in which the drug will be used clinically.
2.
Allows blood levels to be
measured at the same concentrations encountered therapeutically.
3.
Easy to predict the peak and
valley characteristics of drug since the bioavailability is determined at
steady-state.
4.
Requires collection of fewer
blood samples.
5.
The drug blood levels are higher
due to cumulative effect which makes its determination possible even by the
less sensitive analytic methods.
6.
Can be ethically performed in
patients because of the therapeutic benefit to the patient.
7.
Small inter-subject variability
is observed in such a study which allows use of fewer subjects.
8.
Better evaluation of the
performance of a controlled-release formulation is possible.
9.
Nonlinearity in pharmacokinetics,
if present, can be easily detected.
10. Eliminates the need for a long wash-out period between doses. Moreover,
the switch-over from one formulation to the other is possible at steady state.
Limitations of multiple-dose studies include –
1.
Tedious, requires more time to
complete.
2.
More difficult and costly to
conduct (requires prolonged monitoring of subjects).
3.
Poor compliance by subjects.
4.
Greater exposure of subjects to
the test drug, increasing the potential for adverse reactions.
In multiple dose study, one must ensure that the
steady-state has been reached. For this, the drug should be administered for 5
to 6 elimination half-lives before collecting the blood samples.
Human Volunteers—Healthy Subjects versus Patients
Ideally, the bioavailability study should be
carried out in patients for whom the
drug is intended to be used because of apparent advantages—
1.
The patient will be benefited
from the study.
2.
Reflects better the therapeutic
efficacy of a drug.
3.
Drug absorption pattern in
disease states can be evaluated.
4.
Avoids the ethical quandary of
administering drugs to healthy subjects.
Patients are generally preferred in multiple dose
bioavailability studies. The drawbacks of using patients as volunteers are
equally large—disease, other drugs, physiologic changes, etc. may modify the
drug absorption pattern. Stringent study conditions such as fasting state
required to be followed by the subject is also difficult. In short,
establishing a standard set of conditions necessary for a bioavailability study
is difficult with patients as volunteers. Such studies are therefore usually
performed in young (20 to 40 years), healthy, male adult volunteers (body
weight within a narrow range; ± 10%), under restricted dietary and fixed
activity conditions. Female volunteers are used only when drugs such as oral
contraceptives are to be tested. The number of subjects to be selected depends
upon the extent of inter-subject variability but should be kept to a minimum
required to obtain a reliable data. The consent of volunteers must be obtained
and they must be informed about the importance of the study, conditions to be
followed during the study and possible hazards if any, prior to starting the
study. Medical examination should be performed in order to exclude subjects
with any kind of abnormality or disease. The volunteers must be instructed to
abstain from any medication for at least a week and to fast overnight prior to
and for a minimum of 4 hours after dosing. The volume and type of fluid and the
standard diet to be taken must also be specified. Drug washout period for a
minimum of ten biological half-lives must be allowed for between any two
studies in the same subject.
Measurement of Bioavailability
The methods useful in quantitative evaluation of
bioavailability can be broadly divided into two categories — pharmacokinetic methods and pharmacodynamic methods.
I. Pharmacokinetic Methods
These are very widely used and based on the
assumption that the pharmacokinetic profile reflects the therapeutic
effectiveness of a drug. Thus, these are indirect
methods. The two major pharmacokinetic methods are:
1. Plasma level-time studies.
2. Urinary excretion studies.
II. Pharmacodynamic Methods
These methods are complementary to pharmacokinetic
approaches and involve direct
measurement of drug effect on a (patho)physiological process as a function of
time. The two pharmacodynamic methods involve determination of bioavailability
from:
1. Acute pharmacological
response.
2. Therapeutic response.
Plasma Level—Time Studies
Unless determination of plasma drug concentration
is difficult or impossible, it is the most reliable method and method of choice
in comparison to urine data. The method is based on the assumption that two
dosage forms that exhibit superimposable plasma level-time profiles in a group
of subjects should result in identical therapeutic activity.
With single dose study, the method requires
collection of serial blood samples for a period of 2 to 3 biological half-lives
after drug administration, their analysis for drug concentration and making a
plot of concentration versus corresponding time of sample collection to obtain
the plasma level-time profile. With i.v. dose, sampling should start within 5
minutes of drug administration and subsequent samples taken at 15 minute
intervals. To adequately describe the disposition phase, at least 3 sample
points should be taken if the drug follows one-compartment kinetics and 5 to 6
points if it fits two-compartment model. For oral dose, at least 3 points
should be taken on the ascending part of the curve for accurate determination
of Ka. The points for disposition or descending phase of the curve
must be taken in a manner similar to that for i.v. dose.
The 3 parameters of plasma level-time studies which
are considered important for determining bioavailability are:
1. Cmax: The peak plasma concentration that gives an indication whether the drug is sufficiently absorbed
systemically to provide a therapeutic response. It is a function of both the
rate and extent of absorption. Cmax will increase with
an increase in the dose, as well as with an increase in the absorption rate.
2. tmax: The peak time that gives an indication of the rate of absorption. It decreases as the rate of absorption
increases.
3. AUC: The area under the plasma level-time curve that gives a measure of the extent of absorption or the amount of
drug that reaches the systemic circulation.
The extent of bioavailability can be determined by
following equations:
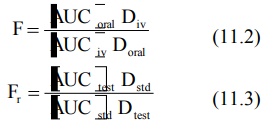
where D stands for dose administered and subscripts
iv and oral indicate the route of administration. Subscripts test and std indicate the test and the standard doses of the same drug to
determine relative availability. The rate of absorption can be computed from
one of the several methods discussed in Chapter
9.
With multiple dose study, the method involves drug
administration for at least 5 biological half-lives with a dosing interval
equal to or greater than the biological half-life (i.e. administration of at
least 5 doses) to reach the steady-state. A blood sample should be taken at the
end of previous dosing interval and 8 to 10 samples after the administration of
next dose. The extent of bioavailability is given as:
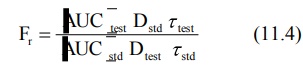
where [AUC] values are area under the plasma
level-time curve of one dosing interval in a multiple dosage regimen, after
reaching the steady-state (Fig. 11.1) and τ is the
dosing interval.
Bioavailability can also be determined from the
peak plasma concentration at steady-state Css,max according to
following equation:
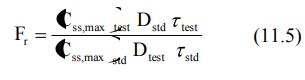
The rate of absorption is not important in the
multiple dosing methods.
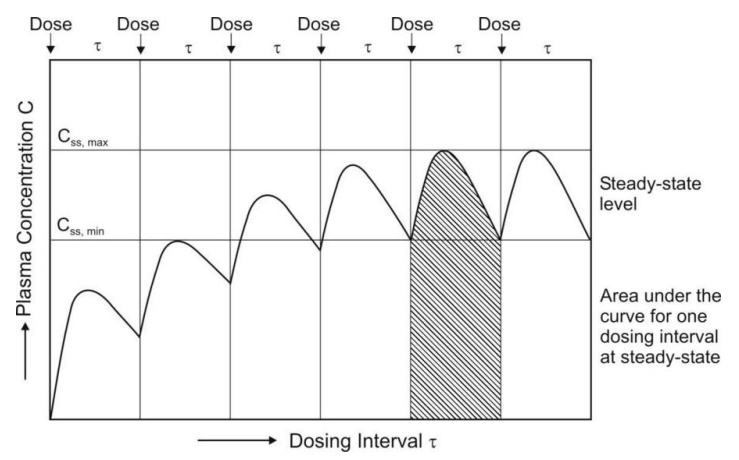
Fig. 11.1 Determination of AUC and Css,max on multiple dosing upto steady-state
Urinary Excretion Studies
This method of assessing bioavailability is based on the principle that the urinary excretion of unchanged drug is directly proportional
to the plasma concentration of drug. As
a rule of thumb, determination of bioavailability using urinary excretion data
should be conducted only if at least 20% of administered dose is excreted
unchanged in the urine. The study is particularly useful for –
·
Drugs extensively excreted
unchanged in the urine – for example, certain thiazide diuretics and
sulphonamides.
·
Drugs that have urine as the site
of action - for example, urinary antiseptics such as nitrofurantoin and
hexamine.
The method has several advantages and disadvantages
as discussed in Chapter 9.
Concentration of metabolites excreted in urine is never taken into account in
calculations since a drug may undergo presystemic metabolism at different
stages before being absorbed. The method involves –
·
Collection of urine at regular
intervals for a time-span equal to 7 biological half-lives.
·
Analysis of unchanged drug in the
collected sample.
·
Determination of the amount of
drug excreted in each interval and cumulative amount excreted.
For obtaining valid results, following criteria
must be met further –
·
At each sample collection, total
emptying of the bladder is necessary to avoid errors resulting from addition of
residual amount to the next urine sample.
·
Frequent sampling of urine is
also essential in the beginning in order to compute correctly the rate of
absorption.
·
The fraction excreted unchanged
in urine must remain constant.
The three major parameters examined in urinary
excretion data obtained with a single dose study are:
1. (dXu/dt)max: The maximum urinary excretion rate, it is
obtained from the peak of plot
between rate of excretion versus midpoint time of urine collection period. It
is analogous to the Cmax
derived from plasma level studies since the rate of appearance of drug in the
urine is proportional to its concentration in systemic circulation. Its value
increases as the rate of and/or extent of absorption increases (see Fig. 11.2).
2. (tu)max: The time for maximum excretion rate, it is
analogous to the tmax of plasma level data. Its
value decreases as the absorption rate increases.
3. Xu ∞ : The cumulative amount of drug
excreted in the urine, it is related to the AUC of plasma level data and increases as the extent of absorption
increases.
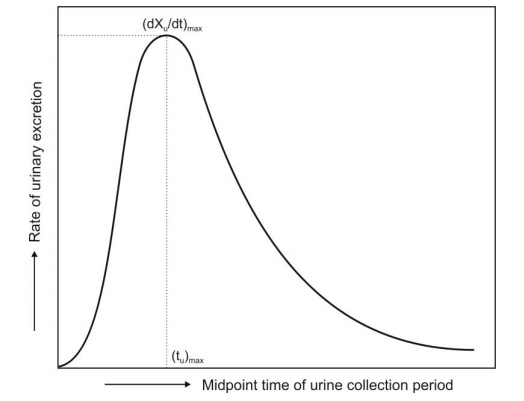
Fig. 11.2 Plot of urinary excretion rate versus time. Note that the curve is
analogous to a typical plasma
level-time profile obtained after oral administration of a single dose of drug.
The extent of bioavailability is calculated from
equations given below:
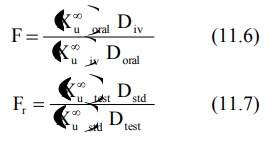
With multiple dose study to steady-state, the
equation for computing bioavailability is:
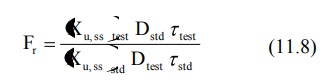
where (Xu,ss) is the amount of drug
excreted unchanged during a single dosing interval at steady-state.
In practice, estimation of bioavailability by
urinary excretion method is subject to a high degree of variability, and is
less reliable than those obtained from plasma concentration-time profiles. It
is thus not recommended as a substitute for blood concentration data; rather,
it should be used in conjunction with blood level data for confirmatory
purposes.
Bioavailability can also be determined for a few
drugs by assay of biologic fluids other than plasma and urine. In case of
theophylline, salivary excretion can be used whereas for cephalosporin
antibiotics, appearance of drug in CSF and bile can be determined. Caution must
however be exercised to account for salivary and enterohepatic cycling of the
drugs.
Acute Pharmacological Response Method
When bioavailability measurement by pharmacokinetic
methods is difficult, inaccurate or non-reproducible, an acute pharmacological
effect such as a change in ECG or EEG readings, pupil diameter, etc. is related
to the time course of a given drug. Bioavailability can then be determined by
construction of pharmacological effect-time curve as well as dose-response
graphs. The method requires measurement of responses for at least 3 biological
half-lives of the drug in order to obtain a good estimate of AUC.
Disadvantages of this method include –
1. The pharmacological response
tends to be more variable and accurate correlation between measured response
and drug available from the formulation is difficult.
2. The observed response may be
due to an active metabolite whose concentration is not proportional to the
concentration of parent drug responsible for the pharmacological effect.
Therapeutic Response Method
Theoretically the most definite, this method is
based on observing the clinical response to a drug formulation given to
patients suffering from disease for which it is intended to be used. However,
the method has several drawbacks –
1. Quantitation of observed
response is too improper to allow for reasonable assessment of relative
bioavailability between two dosage forms of the same drug.
2. Bioequivalence studies are usually conducted
using a crossover design in which each subject receives each of the test dosage
forms, and it is assumed that the physiological status of the subject does not
change significantly over the duration of the study.
3. Unless multiple-dose protocols
are employed, a patient who required the drug for a disease would be able to
receive only a single dose of the drug every few days or perhaps each week.
4. Many patients receive more
than one drug, and the results obtained from a bioavailability study could be
compromised because of a drug–drug interaction.
Because of the above considerations, the general
conclusion is that most bioavailability/bioequivalence studies should be
carried out in healthy subjects. However, for drugs that are not designed to be
absorbed into the systemic circulation and are active at the site of
administration, clinical studies in patients are the only means to determine
bioequivalence. Such studies are usually conducted using a parallel, rather
than a crossover design. Examples include studies of topical antifungal agents,
drugs used in the treatment of acne and agents such as sucralfate used in ulcer
therapy.
In Vitro Drug Dissolution Rate and Bioavailability
The physicochemical property of most drugs that has
greatest influence on their absorption characteristics from the GIT is
dissolution rate. The best way of assessing therapeutic efficacy of drugs with
a slow dissolution rate is in vivo
determination of bioavailability which is usually done whenever a new
formulation is to be introduced into the market. However, monitoring
batch-to-batch consistency through use of such in vivo tests is extremely costly, tedious and time consuming
besides exposing the healthy subjects to hazards of drugs. It would therefore
be always desirable to substitute the in
vivo bioavailability tests with
inexpensive in vitro methods. The
simple in vitro disintegration test
is unreliable. The best available tool
today which can at least quantitatively
assure about the biologic availability of a drug from its formulation is its in vitro dissolution test.
In Vitro Drug Dissolution Testing Models
For an in
vitro test to be useful, it must predict the in vivo behaviour to such an extent that in vivo bioavailability test need not be performed. Despite
attempts to standardize the test
performance, the in vitro dissolution
technique is still by no means a perfect approach. The efforts are mainly aimed
at mimicking the environment offered by the biological system.
There are several factors that must be considered
in the design of a dissolution test. They are –
·
Factors relating to the
dissolution apparatus such as—the design, the size of
the
container (several mL to several litres), the shape of the container
(round bottomed or flat), nature of agitation (stirring, rotating or oscillating
methods), speed of agitation, performance precision of the apparatus, etc.
·
Factors relating to the
dissolution fluid such as—composition (water, 0.1N
HCl,
phosphate buffer, simulated gastric fluid, simulated intestinal fluid,
etc.), viscosity, volume (generally larger than that needed to completely
dissolve the drug under test), temperature (generally 37oC) and
maintenance of sink (drug concentration
in solution maintained constant at a low level) or non-sink conditions (gradual increase in the drug concentration in
the dissolution medium).
·
Process parameters such as method of introduction of dosage form, sampling techniques,
changing the dissolution fluid, etc.
The ideal features of a dissolution
apparatus are:
1.
The fabrication, dimensions, and
positioning of all components must be precisely specified and reproducible,
run-to-run.
2.
The apparatus must be simple in
design, easy to operate and useable under a variety of conditions.
3.
The apparatus must be sensitive
enough to reveal process changes and formulation differences but still yield
repeatable results under identical conditions.
4.
The apparatus, in most cases,
should permit controlled variable intensity of mild, uniform, non-turbulent
liquid agitation.
5.
Nearly perfect sink conditions
should be maintained.
6.
The apparatus should provide an
easy means of introducing the dosage form into the dissolution medium and
holding it, once immersed, in a regular reliable fashion.
7.
The apparatus should provide
minimum mechanical abrasion to the dosage form during the test period to avoid
disruption of the microenvironment surrounding the dissolving form.
8.
Evaporation of the solvent medium
must be eliminated, and the medium must be maintained at a fixed temperature
within a specified narrow range. Most apparatuses are thermostatically
controlled at around 37°C.
9.
Samples should be easily
withdrawn for automatic or manual analysis without interrupting the flow
characteristics of the liquid.
10. The apparatus should be capable of allowing the evaluation of
disintegrating, non-disintegrating, dense or floating tablets or capsules, and
finely powdered drugs.
11. The apparatus should allow good inter-laboratory agreement.
The dissolution apparatus has evolved gradually and
considerably from a simple beaker type to a highly versatile and fully automated
instrument. The devices can be classified in a number of ways. Based on the
absence or presence of sink conditions, there are two principal types of dissolution apparatus:
1. Closed-compartment apparatus: It is
basically a limited-volume apparatus operating
under non-sink conditions. The dissolution fluid is restrained to the size of
the container, e.g. beaker type apparatuses such as the rotating basket and the
rotating paddle apparatus.
2. Open-compartment (continuous flow-through) apparatus: It is the one in which the
dosage form is contained in a column which is brought in continuous contact
with fresh, flowing dissolution medium (perfect sink condition).
A third type called as dialysis systems are used for very poorly aqueous soluble drugs for
which maintenance of sink conditions would otherwise require large volume of dissolution
fluid. Only the official or compendial methods (USP methods) will be discussed
here briefly.
Rotating Basket Apparatus (Apparatus 1)
First described by Pernarowski et al, it is basically a closed-compartment, beaker type apparatus
comprising of a cylindrical glass vessel with hemispherical bottom of one litre
capacity partially immersed in a water bath to maintain the temperature at 37oC.
A cylindrical basket made of 22 mesh to hold the dosage form is located
centrally in the vessel at a distance of 2 cm from the bottom and rotated by a
variable speed motor through a shaft (Fig. 11.3a). The basket should remain in motion during drawing of samples.
All metal parts like basket and shaft are made of SS 316.
Rotating Paddle Apparatus (Apparatus 2)
The assembly is same as that for apparatus 1 except
that the rotating basket is replaced with a paddle which acts as a stirrer
(Fig. 11.3b). The method was first
described by Levy and Hayes. The dosage form is allowed to sink to the bottom
of the vessel. Sinkers are recommended to prevent floating of capsules and
other floatable forms. A small, loose, wire helix may be attached to such
preparations to prevent them from floating.
Reciprocating Cylinder Apparatus (Apparatus 3)
This apparatus consists of a set of cylindrical
flat-bottomed glass vessels equipped with reciprocating cylinders (Fig. 11.3c). The apparatus is particularly used
for dissolution testing of controlled release bead-type (pellet) formulations.
Flow-Through Cell Apparatus (Apparatus 4)
The flow-through apparatus consists of a reservoir
for the dissolution medium and a pump that forces dissolution medium through
the cell holding the test sample. It may be used in either –
·
Closed-mode where the fluid is
recirculated and, by necessity, is of fixed volume, or
·
Open-mode when there is
continuous replenishment of the fluids.
The material under test (tablet, capsules, or
granules) is placed in the vertically mounted dissolution cell, which permits
fresh solvent to be pumped (between 240 and 960 mL/h) in from the bottom (Fig.
11.3d). Advantages of this apparatus
include –
1. Easy maintenance of sink
conditions for dissolution which is often required for drugs having limited aqueous
solubility.
2. Feasibility of using large
volume of dissolution fluid.
3. Feasibility for automation of
apparatus.
Paddle Over Disc Apparatus (Apparatus 5)
This apparatus is used for evaluation of
transdermal products and consists of a sample holder or disc that holds the
product. The disc is placed at the bottom of apparatus 2 (rotating paddle
apparatus; see fig. 11.3e) and the
apparatus operated in the usual way.
Cylinder Apparatus (Apparatus 6)
This apparatus is also used for evaluation of
transdermal products and is similar to apparatus 1 (Fig. 11.3f). Instead of basket, a stainless steel
cylinder is used to hold the sample. The sample is mounted on an inert porous
cellulosic material and adhered to the cylinder.
Reciprocating Disc Apparatus (Apparatus 7)
This apparatus is used for evaluation of
transdermal products as well as non-disintegrating controlled-release oral
preparations. The samples are placed on disc-shaped holders (Fig. 11.3g) using inert porous cellulosic support
which reciprocates vertically by means of a drive inside a glass container
containing dissolution medium. The test is carried out at 320C and
reciprocating frequency of 30 cycles/min.
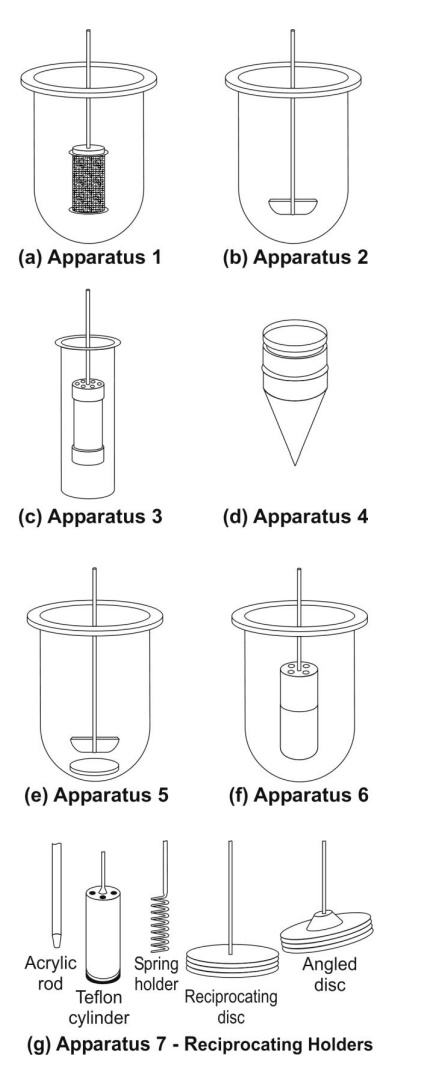
Fig. 11.3 Schematic representation of official USP dissolution apparatus - (a)
Apparatus 1 - rotating basket apparatus, (b) Apparatus 2 - rotating paddle apparatus,
(c) Apparatus 3 – reciprocating cylinder apparatus, (d) Apparatus 4 – flow
through cell apparatus, (e) Apparatus 5 – paddle over disc apparatus, (f)
Apparatus 6 – cylinder apparatus, and (g) Apparatus 7 – reciprocating disc
apparatus
Table 11.1 lists the various types of dissolution
apparatus and their applications, and table 11.2 summarises the dissolution
methodology to be adopted for immediate-release products on the basis of BCS.
Table 11.1.
Compendial Dissolution Apparatus Types and Their Applications
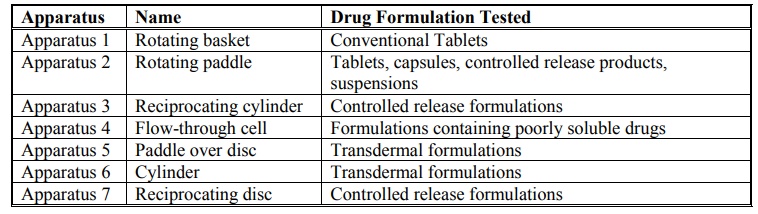
Table 11.2.
Dissolution Methodology for Immediate-Release Products Based on BCS

Dissolution Acceptance Criteria
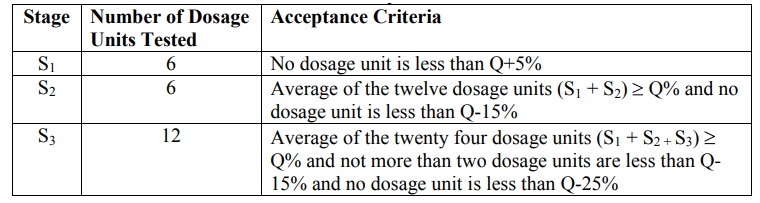
On the basis of dissolution profile data, criteria
for acceptance/passing of test results are based on Q values as given in table
11.3. The value of Q is defined as
percentage of drug content dissolved
in a given time period. This value is generally specified in USP monograph of a given drug product.
Three stages viz. S1, S 2
and S3 of dissolution testing are allowed as given in table 11.3.
In the first stage of the USP dissolution test
consists of testing six dosage units. If all of the dosage units are greater
than or equal to Q+5%, then the dissolution test criteria are met and the test
is passed. However, if this criterion is not met, six additional dosage units
are tested and compared to the acceptance criteria for the twelve dosage units.
To pass at the second stage, the average of the twelve dosage units must be
equal to or greater than Q and no dosage unit can be less than Q-15%. If both
of the above criteria are not met at the second stage, the final stage of
testing is performed. Twelve additional dosage units are evaluated, providing a
total of twenty four results. To pass at this final stage of testing, the
average of the twenty four dosage units must be equal to or greater than Q, not
more than two dosage units can be less than Q - 15 %, and no dosage unit can be
less than Q-25%.
Table 11.3.
Dissolution Acceptance Criteria
Objectives of Dissolution Profile Comparison
Comparison of in
vitro dissolution profiles of test drug product and approved drug product are
useful for –
·
Development of bioequivalent drug
products.
·
Demonstrating equivalence after
change in formulation of drug product.
·
Biowaiver of drug product of
lower dose strength in proportion to higher dose strength drug product
containing same active ingredient and excipients.
Method for Comparison of Dissolution Profile
A model
independent method for comparison of two dissolution profiles is based on
determination of difference factor f1 and similarity factor f2
which are calculated using following formulae –
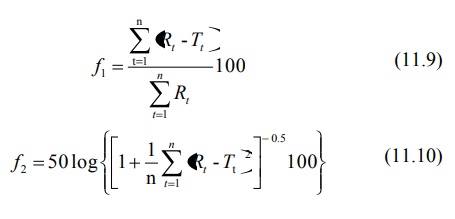
where
n = number of dissolution time points
Rt = dissolution value of the
reference drug product at time t
Tt = dissolution value of the test
drug product at time t
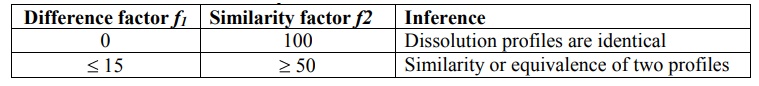
The guidelines adopted for interpreting f1
and f2
values
are given in table 11.4.
Table 11.4.
Comparison of Dissolution Profile
The evaluation of similarity between dissolution
profiles is based on following conditions –
·
Minimum of three dissolution time
points are measured.
·
Number of drug products tested
for dissolution is 12 for both test and reference.
·
Not more than one mean value of
> 85% dissolved for each product.
·
Standard deviation of mean of any
product should not be more than 10% from second to last dissolution time point.
In Vitro—In Vivo Correlation (IVIVC)
A simple in
vitro dissolution test on the drug product will be insufficient to predict
its therapeutic efficacy. Convincing correlation between in vitro dissolution behaviour of a drug and its in vivo bioavailability must be
experimentally demonstrated to guarantee reproducibility of biologic response. In
vitro-in vivo correlation is
defined as the predictive
mathematical model that describes the relationship between an in-vitro property
(such as the rate and extent of dissolution) of a dosage form and an in-vivo
response (such as the plasma drug concentration or amount of drug absorbed).
The main objective of developing and evaluating an
IVIVC is to enable the dissolution test to serve as a surrogate (alternate) for in vivo bioavailability studies in human beings.
The applications of developing such an
IVIVC are —
1. To ensure batch-to-batch
consistency in the physiological performance of a drug product by use of such in vitro values.
2. To serve as a tool in the
development of a new dosage form with desired in vivo performance.
3. To assist in validating or
setting dissolution specifications (i.e. the dissolution specifications are
based on the performance of product in
vivo).
There are two basic approaches by which a
correlation between dissolution testing and bioavailability can be developed:
1. By establishing a
relationship, usually linear, between the in
vitro dissolution and the in vivo
bioavailability parameters.
2. By using the data from
previous bioavailability studies to modify the dissolution methodology in order
to arrive at meaningful in vitro-in vivo
correlation.
Though the former approach is widely used, the
latter still holds substance, since to date, there is no single dissolution
rate test methodology that can be applied to all drugs.
Some of the often used quantitative linear in vitro-in vivo correlations are –
1. Correlations Based on the Plasma Level Data: Here linear relationships between
dissolution parameters such as percent drug dissolved, rate of dissolution,
rate constant for dissolution, etc. and parameters obtained from plasma level
data such as percent drug absorbed, rate of absorption, Cmax, tmax,
Ka, etc. are developed; for example, percent drug dissolved versus
percent drug absorbed plots.
2. Correlation Based on the Urinary Excretion Data: Here, dissolution parameters
are correlated to the amount of drug excreted unchanged in the urine,
cumulative amount of drug excreted as a function of time, etc.
3. Correlation Based on the Pharmacological Response: An acute pharmacological
effect such as LD50 in animals is related to any of the dissolution
parameters.
Statistical moments theory can also
be used to determine the relationship such as mean dissolution time (in
vitro) versus mean residence time (in
vivo).
Though examples of good correlations are many,
there are instances when positive correlation is difficult or impossible; for
example, in case of corticosteroids, the systemic availability may not depend
upon the dissolution characteristics of the drug. Several factors that limit
such a correlation include variables pertaining to the drug such as dissolution
methodology, physicochemical properties of the drug such as particle size,
physiologic variables like presystemic metabolism, etc.
In vitro-In vivo Correlation Levels
Three IVIVC levels have been defined and
categorised in descending order of usefulness.
Level A – The highest category of
correlation, it represents a point-to-point
relationship between in vitro
dissolution and the in vivo rate of
absorption (or in vivo dissolution) i.e. the in vitro dissolution and in vivo absorption rate curves are superimposable and the mathematical
description for both curves is the same.
Advantages of level A correlation are as follows –
1. A point-to-point correlation
is developed. The in vitro
dissolution curve serves as a surrogate for in
vivo performance. Any change in manufacturing procedure or modification in
formula can be justified without the need for additional human studies.
2. The in vivo dissolution serves as in
vivo indicating quality control procedure for predicting dosage form‘s
performance.
Level B – Utilises the principles of
statistical moment analysis. The mean in vitro dissolution time is compared to either the mean residence time or
the mean in vivo dissolution time.
However, such a correlation is not a point-to-point correlation since there are
a number of in vivo curves that will
produce similar mean residence time values. It is for this reason that one
cannot rely upon level B correlation to justify changes in manufacturing or
modification in formula. Moreover, the in
vitro data cannot be used for quality control standards.
Level C – It is a single point correlation.
It relates one dissolution time point (e.g.
t50%, etc.) to one pharmacokinetic parameter such as AUC, tmax
or C max. This level is generally useful only as a guide in
formulation development or quality control owing to its obvious limitations.
Multiple Level C – It is correlation involving one
or several pharmacokinetic parameters
to the amount of drug dissolved at various time points.
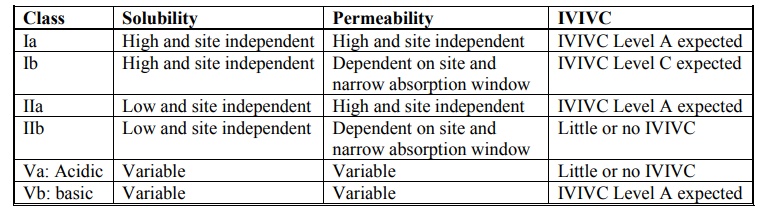
Biopharmaceutics Classification System (BCS) and In vitro-In vivo Correlation (IVIVC)
The Biopharmaceutics Classification System (BCS) is
a fundamental guideline for determining the conditions under which in-vitro in-vivo correlations are
expected. Table 11.5 indicates whether IVIVC is expected or possible for
various drug categories when formulated as controlled-release preparations. The
importance of BCS in formulation design and drug delivery is further
highlighted in table 11.8.
Table 11.5.
Biopharmaceutics Drug Classification System for Extended Release Drug
Products
BCS-Based Biowaiver to In Vivo Bioavailability/Bioequivalence Studies
According to BCS, in vivo bioavailability and bioequivalence studies need not be
conducted for drug products under following circumstances -
·
Rapid and similar dissolution.
·
High solubility.
·
High permeability.
·
Wide therapeutic window.
·
Excipients used in dosage form
are same as those present in approved drug product.
Related Topics
