Bioequivalence Studies
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Bioavailability and Bioequivalence
If a new product is intended to be a substitute for an approved medicinal product as a pharmaceutical equivalent or alternative, the equivalence with this product should be shown or justified.
BIOEQUIVALENCE STUDIES
Need/Objectives for Biequivalence Studies
If a new product is intended to be a substitute for
an approved medicinal product as a pharmaceutical equivalent or alternative,
the equivalence with this product should be shown or justified. In order to
ensure clinical performance of such drug products, bioequivalence studies
should be performed. Bioequivalence studies are conducted if there is:
·
A risk of bio-inequivalence
and/or
·
A risk of pharmacotherapeutic
failure or diminished clinical safety.
Some of the important terms relevant in this
context will be defined.
Equivalence: It is a relative term that
compares drug products with respect to a
specific characteristic or function or to a defined set of standards. There
are several types of equivalences.
Chemical Equivalence: It
indicates that two or more drug products contain the same labelled chemical substance as an active ingredient in the same
amount.
Pharmaceutical Equivalence: This term
implies that two or more drug products are
identical in strength, quality, purity, content uniformity and
disintegration and dissolution characteristics; they may however differ in
containing different excipients.
Bioequivalence: It is a relative term which denotes
that the drug substance in two or more
identical dosage forms, reaches the systemic circulation at the same relative
rate and to the same relative extent i.e. their plasma concentration-time
profiles will be identical without significant statistical differences.
When statistically significant differences are
observed in the bioavailability of two or more drug products, bio-inequivalence is indicated.
Therapeutic Equivalence: This term
indicates that two or more drug products that contain the same therapeutically active ingredient elicit
identical pharmacological effects and can control the disease to the same
extent.
Types of Bioequivalence Studies
Bioequivalence can be demonstrated either –
·
In vivo, or
·
In vitro.
In vivo Bioequivalence Studies
The following sequence of criteria is useful in
assessing the need for in vivo
studies:
1. Oral immediate release products with systemic action
·
Indicated for serious conditions
requiring assured response
·
Narrow therapeutic margin
·
Pharmacokinetics complicated by
absorption < 70% or absorption window, nonlinear kinetics, presystemic
elimination > 70%
·
Unfavourable physiochemical
properties, e.g. low solubility, metastable modifications, instability, etc.
·
Documented evidence for
bioavailability problems
·
No relevant data available,
unless justification by applicant that in
vivo study is not necessary.
2. Non-oral immediate release products.
3. Modified release products with systemic action.
In vivo bioequivalence studies are
conducted in the usual manner as discussed for bioavailability studies, i.e. the pharmacokinetic and the
pharmacodynamic methods.
In vitro Bioequivalence Studies
If none of the above criteria is applicable,
comparative in vitro dissolution
studies will suffice. In vitro
studies, i.e. dissolution studies can be used in lieu of in vivo bioequivalence under certain circumstances, called as biowaivers
(exemptions) –
1. The drug product differs only in strength of the
active substance it contains, provided all the following conditions hold –
·
Pharmacokinetics are linear
·
The qualitative composition is
the same
·
The ratio between active
substance and the excipients is the same, or (in the case of small strengths)
the ratio between the excipients is the same
·
Both products are produced by the
same manufacturer at the same production site
·
A bioavailability or
bioequivalence study has been performed with the original product
·
Under the same test conditions,
the in vitro dissolution rate is the
same.
2. The drug product has been
slightly reformulated or the manufacturing method has been slightly modified by
the original manufacturer in ways that can convincingly be argued to be
irrelevant for the bioavailability.
3. The drug product meets all of
the following requirements –
·
The product is in the form of
solution or solubilised form (elixir, syrup, tincture, etc.)
·
The product contains active
ingredient in the same concentration as the approved drug product.
·
The product contains no
excipients known to significantly affect absorption of the active ingredient.
4. An acceptable IVIVC and the in vitro dissolution rate of the new
product is equivalent with that of the already approved medicinal product.
Moreover,
·
The product is intended for
topical administration (cream, ointment, gel, etc.) for local effect.
·
The product is for oral
administration but not intended to be absorbed (antacid or radio-opaque
medium).
·
The product is administered by
inhalation as a gas or vapour.
The criteria for drug products listed above
indicate that bioavailability and bioequivalence are self-evident.
Bioequivalence Experimental Study Design
The various types of test designs that are usually
employed in clinical trials, bioavailability and bioequivalence studies are
discussed below.
1. Completely randomised designs
In a completely randomised design, all treatments (factor levels) are
randomly allocated among all experimental subjects.
Method of randomisation
Label all subjects with the same number of digits,
for e.g., if there are 20 subjects, number them from 1 to 20. Randomly select
non-repeating random numbers (like simple randomisation) with among these
labels for the first treatment, and then repeat for all other treatments.
Advantages
1) The design is extremely easy
to construct.
2) It can accommodate any number of treatments and
subjects.
3) The design is easy and simple
to analyse even though the sample sizes might not be the same for each
treatment.
Disadvantages
1) Although the design can be
used for any number of treatments, it is best suited for situations in which
there are relatively few treatments.
2) All subjects must be as
homogeneous as possible. Any extraneous sources of variability will tend to
inflate the random error term, making it difficult to detect differences among
the treatment (or factor level) mean responses.
2. Randomised block designs
First, subjects are sorted into homogeneous groups,
called blocks and the treatments are then assigned at random within the blocks.
Method of Randomisation
Subjects having similar background characteristics
are formed as blocks. Then treatments are randomised within each block, just
like the simple randomisation. Randomisations for different blocks are done
independent of each other.
Advantages
1.
With effective and systematic way
of grouping, it can provide substantially more precise results than a
completely randomised design of comparable size.
2.
It can accommodate any number of
treatments or replications.
3.
Different treatments need not
have equal sample size.
4.
The statistical analysis is
relatively simple. The design is easy to construct.
5.
If an entire treatment or block
needs to be dropped from the analysis for some reason, such as spoiled results,
the analysis is not thereby complicated.
6.
Variability in experimental units
can be deliberately introduced to widen the range of validity of the experimental
results without sacrificing the precision of results.
Disadvantages
1.
Missing observations within a
block require more complex analysis.
2.
The degrees of freedom of
experimental error are not as large as with a completely randomised design.
3. Repeated measures, cross-over and carry-over designs
This is essentially a randomised block design in which the same subject
serves as a block. The same subject is utilized for
each of the treatments under study. Since we take repeated measures on each
subject we get the design name ―repeated measures design‖. The study may
involve several treatments or a single treatment evaluated at different points
in time. The administration of two or
more treatments one after the other in a specified or random order to the same group of patients is called a crossover design or change-over design. The drawback of
crossover studies is the potential for distortion due to carry-over, that is,
residual effects from preceding treatments. To prevent carry-over effects,
one must always allow for a wash-out period during which most of the
drug is eliminated from the body – generally about 10 elimination
half-lives. Example: clinical trials to monitor safety and side effects.
Method of Randomisation
Complete randomisation is used to randomise the
order of treatments for each subject.
Randomisations for different subjects are
independent of each other.
Advantages
·
They provide good precision for
comparing treatments because all sources of variability between subjects are
excluded from the experimental error.
·
It is economic on subjects. This
is particularly important when only a few subjects can be utilized for the
experiments.
·
When the interest is in the
effects of a treatment over time, it is usually desirable to observe the same
subject at different points in time rather than observing different subjects at
the specified points in time.
Disadvantages
·
There may be an order effect,
which is connected with the position in the treatment order.
·
There may be a carry-over effect,
which is connected with the preceding treatment or treatments.
4. Latin square designs
Completely randomised design, randomised block
design and repeated measures design are experiments where the
person/subject/volunteer remains on the treatment from the start of the
experiment until the end and thus are called as continuous trial. In a
Latin square, however, each subject receives each treatment during the course
of the experiment. A Latin square design
is a two-factor design (subjects and treatments are the two factors) with one observation in each
cell. Such a design is useful compared the earlier ones when three or more treatments are to be compared and
carry-over effects are balanced. In a Latin square design, rows represent subjects, and columns represent treatments. A r x r
Latin square design is a square with r rows and r columns
such that each of the r2 cells
contain one and only one of the r
letters representing the treatments, and each letter appears once and only once
in ever row and every column. A Latin
square is called standard if the first row and the first column consist of the r
letters in alphabetical order.
Randomised, balanced, cross-over Latin square
design are commonly used for bioequivalence studies.
Advantages
·
It minimizes the inter-subject
variability in plasma drug levels.
·
Minimizes the carry-over effects
which could occur when a given dosage form influences the bioavailability of a
subsequently administered product (intra-subject variability).
·
Minimizes the variations due to
time effect.
·
Treatment effects can be studied
from a small-scale experiment. This is particularly helpful in preliminary or
pilot studies.
·
Makes it possible to focus more
on the formulation variables which is the key to success for any bioequivalence
study.
Disadvantages
·
The use of Latin square design
will lead to a very small number of degrees of freedom for experimental error
when only a few treatments are studied. On the other hand, when many treatments
are studied, the degrees of freedom for experimental error maybe larger than
necessary.
·
The randomisation required is
somewhat more complex than that for earlier designs considered.
·
The study takes a long time since
an appropriate washout period between two administrations is essential which
may be very long if the drug has a long t½.
·
When the number of formulations
to be tested is more, the study becomes more difficult and subject dropout
rates are also high. This can be overcome by use of a balanced incomplete block
design in which a subject receives no more than 2 formulations.
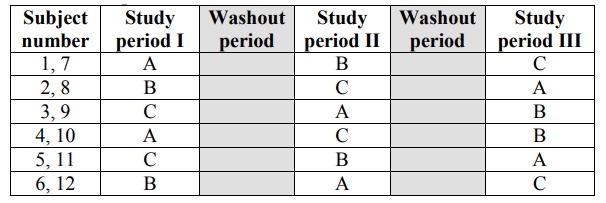
An example of a typical Latin square design is
given in table 11.6.
Table 11.6.
Latin Square Cross-over Design for 6 (or 12) Subjects to Compare Three
Different Formulations, A, B and C
Bioequivalence Study Protocol
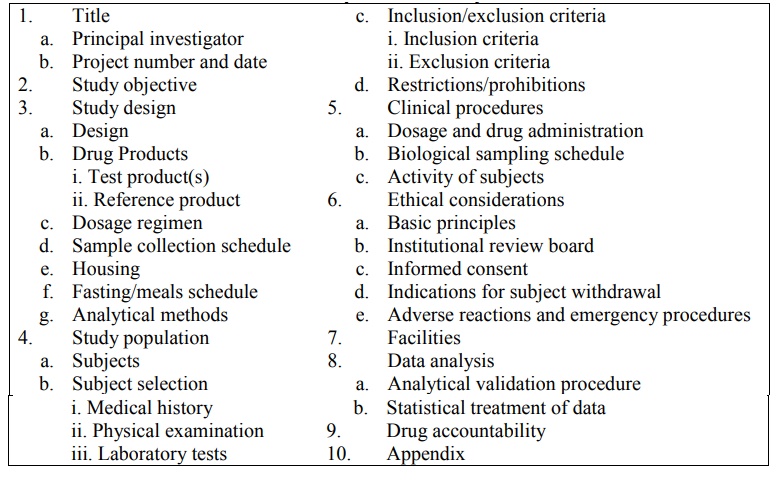
The elements of in
vivo bioequivalence study protocol are listed in table 11.7.
TABLE 11.7.
Elements of Bioequivalence Study Protocol
The in vivo
bioequivalence study requires determination of relative bioavailability after
administration of a single dose of test and reference formulations by the same
route, in equal doses, but at different times. The reference product is
generally a previously approved product, usually the innovator‘s product or
some suitable reference standard. The study is performed in fasting, young,
healthy, adult male volunteers to assure homogeneity in the population and to
spare the patients, elderly or pregnant women from rigors of such a clinical
investigation. Homogeneity in the study population permits focus on formulation
factors.
As for bioavailability studies, either plasma level
or urinary excretion studies may be performed to assess bioequivalence between
drug products. In vitro-in vivo
correlation can also be established for the formulations.
It is always easier to establish bioequivalence
between existing drug products than determination of pharmacokinetics of a new
drug or bioavailability of a new dosage form since —
1. The human volunteers used for
the study of both products are same and all pharmacokinetic parameters can be
assumed to be same for both drug formulations and there is no need to
investigate nonlinearity.
2. The study protocol for all
subjects is uniform, the efficiency of drug absorption from both formulations
can be considered as same and thus differences in absorption pattern can be
ascribed to differences in drug release from the two dosage form.
Statistical Interpretation of Bioequivalence Data
After the data has been collected, statistical
methods must be applied to determine the level of significance of any observed
difference in the rate and/or extent of absorption in order to establish
bioequivalence between two or more drug products. The commonly adopted
approaches to determine statistical differences are –
1. Analysis of variance (ANOVA)
is a statistical procedure used to test the data for differences within and between treatment and control groups. A
statistical difference between the pharmacokinetic parameters obtained from two
or more drug products is considered statistically significant if there is a
probability of less than 1 in 20 or 0.05 (p
≤ 0.05).
The probability p is used to indicate
the level of statistical significance. If p
≤ 0.05, the differences between the two drug products are not considered
statistically significant.
2. Confidence interval approach – Also called as two one-sided test procedure,
it is used to demonstrate if the
bioavailability from the test product is too low or high in comparison to the
reference product. The 90% confidence limits are estimated for the sample means
based on Student’s t distribution of data. A 90% confidence interval
about the ratio of means of the two drug products must be within ±20% for bioavailability parameters such AUC or Cmax i.e. the
difference between the bioavailabilities of the test product should not be
greater than ± 20% of the average of reference product (between 80 and 120%).
When log transformed data are used, the 90% confidence interval is set at
80-125%. These confidence limits are also termed as bioequivalence interval.
Related Topics
