Diastereoselection in Aldol Reactions
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Carbon-Carbon Bond Formation Between Carbon Nucleophiles and Carbon Electrophiles
The reaction of enolates with aldehydes or ketones to produce β-hydroxy car-bonyl derivatives is a very common and a very useful way to make carbon–carbon bonds.
DIASTEREOSELECTION IN ALDOL
REACTIONS
The
reaction of enolates with aldehydes or ketones to produce β-hydroxy car-bonyl derivatives is a very common and a very useful
way to make carbon–carbon bonds. A fundamental stereochemical feature of the
reaction is that two new chiral centers are produced from achiral starting
materials. Hence syn and anti diastereomers will be produced, each as a pair of
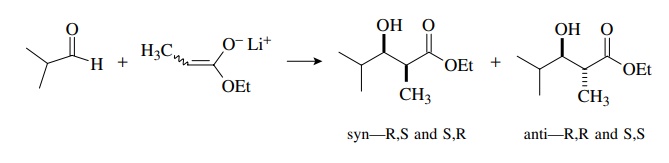
enantiomers. This is shown schematically for the reaction of a propionate
enolate with isobutyraldehyde. Because they have different energies, the syn
and anti diastereomers will be produced in unequal amounts, but each will be
produced in racemic form because both starting materials are achiral.
The
diastereoselectivity of the reaction results from a combination of three
factors. First the carbonyl electrophile can undergo addition on either its Re
or Si face. Second, the enolate nucleophile is planar and can attack the
carbonyl group from either of its faces. Third, the enolate geometry can be
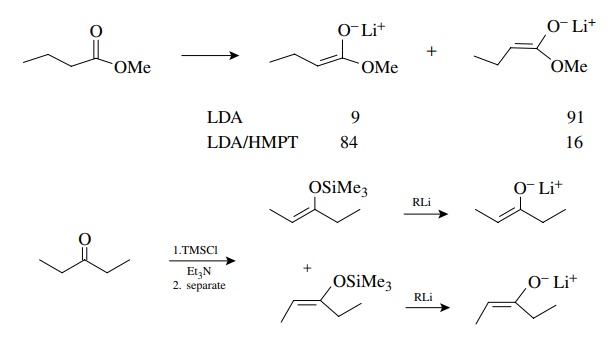
either Z or E. To control the diastereoselectivity, it is first necessary to
use a single isomer of the enolate. In general, the E enolate is the kinetic
enolate and the Z enolate is thermodynamically favored. Methods are available
to produce either as the major isomer by α-proton
removal from carbonyl compounds with strong bases. This is particularly true of
esters and amides. Pure Z and E enolates can also be prepared by first
converting the carbonyl compound to a Z and E mixture of silyl enol ethers,
separating these isomers, and regenerating the Z and E enolates with methyl
lithium. Suffice it to say that there are known ways to produce either Z or E
enolates in pure form.
The
stereoelectronic requirements for carbonyl addition are that electron dona-tion
occurs by interaction of the donor with the π
∗ orbital of the carbonyl group. To
meet the stereoelectronic requirements and explain the diastereoselectivity,
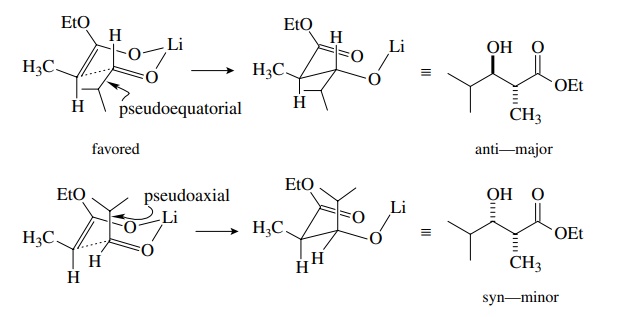
the Zimmerman–Traxler model is used. Interaction of the lithium cation with the
oxygen of the enolate and of the carbonyl electrophile leads to a six-membered
chairlike transition state. If the geometry of the enolate is fixed, the only
variable is the orientation of the electrophile. The preferred orientation has
the larger sub-stituent in a pseudoequatorial position. This preferred orientation
produces the major diastereomer. An example is shown for the Z enolate of ethyl
propionate reacting with isobutyraldehyde, which predicts that the anti
diastereomer should be favored (and it is!). A similar analysis predicts that
the E enolate should give the syn diastereomer as the major product (and it
does!).
This
model is extremely useful in understanding the stereochemical outcomes of aldol
processes. It also provides a framework for influencing the
diastereose-lectivity in a rational way. For instance, if the ethoxy group in
the above example is changed to a much bulkier group, increased transannular
interactions in the pseudoaxial transition state would make it even higher in
energy and result in increased selectivity for the anti isomer (and it does!)
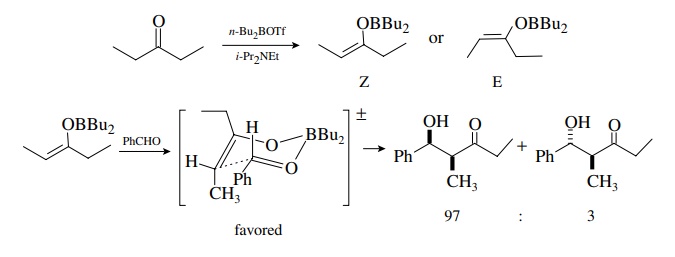
Even
greater diastereoselectivity in the aldol reaction can be achieved using boron
enolates as the carbon nucleophile. Boron enolates are easily prepared from
aldehydes and ketones, and the syn and the anti isomers can be separated as
pure compounds. They react with aldehydes and ketones to give aldol products by
a similar transition state. The difference is that boron oxygen bonds are
shorter than lithium oxygen bonds, and thus steric interactions in the
transition state are magnified and result in greater diastereoselectivity.
Related Topics
