C=C Formation
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Carbon-Carbon Bond Formation Between Carbon Nucleophiles and Carbon Electrophiles
In addition to connecting skeletal fragments by formation of carbon–carbon single bonds, it is also possible to utilize reactions which give carbon–carbon double bonds to assemble carbon skeletons.
C=C FORMATION
In
addition to connecting skeletal fragments by formation of carbon–carbon single
bonds, it is also possible to utilize reactions which give carbon–carbon double
bonds to assemble carbon skeletons. It should be recognized that while the
final products of such reactions contain a carbon–carbon double bond, they are
gen-erally sequential processes in which a single carbon–carbon bond is formed
first and the π bond is formed in a
subsequent elimination step.
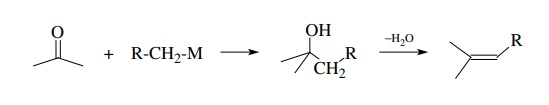
An
elementary example of this process is the reaction of an organometallic
reactant with a ketone (or aldehyde) followed by dehydration of the resulting
alcohol to the olefin. This is truly a sequential process in that the product
alco-hol is dehydrated in a second, independent reaction step. It suffers as a
useful synthetic method because regioisomers are often formed in the
elimination step.
Alternatively
it is possible to have both steps, addition and elimination, occur
spontaneously if appropriate reagents are employed. There are two common
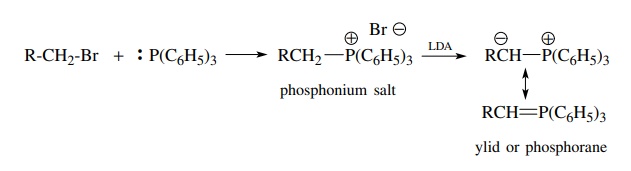
strategies in use: the Wittig reaction and the Wittig–Horner reaction. The
Wittig olefination uses a phosphorus-stabilized carbanion (ylid) as a
nucleophile and a carbonyl compound as an electrophile. Typically the ylid is
generated in situ from a triphenylphosphonium salt and a strong base such as
LDA or an alkyl lithium.
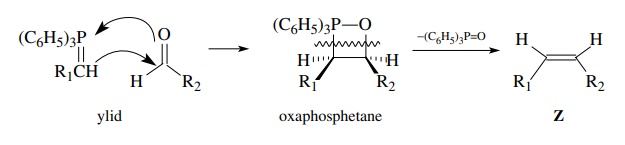
The
ylid is a neutral compound which is resonance stabilized by phosphorus. The
phosphorus atom, being a second-row element, has unfilled d orbitals in the
valence shell that can accept electrons from carbon. Consequently a major
resonance contributor is a structure without formal charges which has a
car-bon–phosphorus double bond. Nevertheless in the resonance hybrid the carbon
atom next to the phosphorus is electron rich and is a good carbon nucleophile
which can add to carbonyl groups to form new carbon–carbon bonds. The cyclic
intermediate (oxaphosphetane) spontaneously loses triphenylphosphine oxide at
room temperature to give an olefin.
In
sum, a new olefinic link is produced, but by an addition–elimination sequence.
In this reaction a stronger C–O double bond in the starting material is
replaced by a weaker C–C double bond in the product. The thermodynamic driving
force for the reaction is the formation of the P–O bond, which is very strong.
The
Wittig reaction is a very important method for olefin formation. The stereochemistry
about the new carbon–carbon double bond is the Z (or less stable) isomer. This unusual stereoselectivity indicates
that product formation is dominated by kinetic control during formation of the
oxaphosphetane.
By
adding a strong base to the cold solution of the oxaphosphetane before it
eliminates, the oxaphosphetane equilibrates to the more stable anti isomer and
the E olefin is produced upon elimination. This so-called Schlosser
modification in conjunction with the normal Wittig reaction enables either the
Z or E isomer of the olefin to be prepared selectively.
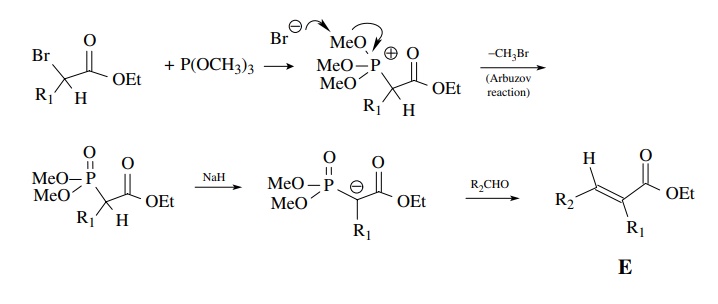
The
Wittig–Horner reaction is the Wittig process applied to carbonyl-activated
ylids and uses trimethylphosphite as the phosphorous reagent. Reaction with a
bromoester gives a phosphate intermediate. Deprotonation with a base such as
sodium hydride and addition of an aldehyde or ketone gives, after elimination
of a phosphonate, an α,β-unsaturated ester. In this case the
intermediate betaine is acidic and undergoes equilibration prior to elimination
so that only the more stable E regioisomer is produced.
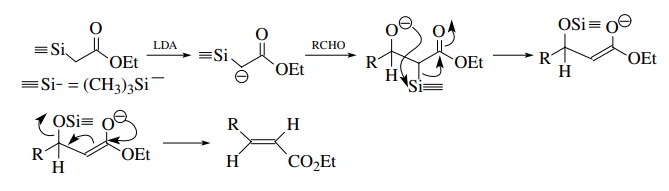
A
recent alternative to the Wittig reaction uses silicon as the atom which
promotes oxygen loss. This reaction, called Peterson olefination, uses an α-silyl anion as the carbon nucleophile
and a carbonyl compound (aldehyde or ketone) is the electrophile. Thus ethyl α-trimethylsilylacetate can be converted
to an enolate and reacts with an aldehyde to give an α,β-unsaturated ester.
The driving force for elimination is the formation of an extremely strong
silicon–oxygen bond, which converts the oxygen atom into a much better silyloxy
leaving group. Only the more stable olefin isomer is produced since
equilibration occurs in the enolate intermediate.
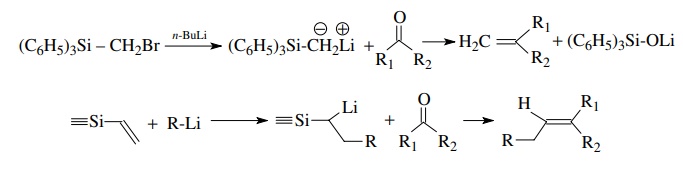
Another
common α-silyl anion is produced by
the halogen exchange from a methyl (but not other group) attached to silicon.
Other α-silyl carbanions can be
generated by other processes. Such anions lack the resonance stabilization of
an ester group seen in the previous example. They are consequently less stable
and must be generated under carefully controlled conditions. They are good
nucleophiles and add effectively to aldehydes and ketones.
Related Topics
