Enolate Regioisomers
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Carbon-Carbon Bond Formation Between Carbon Nucleophiles and Carbon Electrophiles
Enolates are commonly used as the nucleophilic component in carbon–carbon bond-forming reactions. By using strong, nonnucleophilic bases, both esters and ketones are easily converted to their enolates.
ENOLATE REGIOISOMERS
Enolates
are commonly used as the nucleophilic component in carbon–carbon bond-forming
reactions. By using strong, nonnucleophilic bases, both esters and ketones are
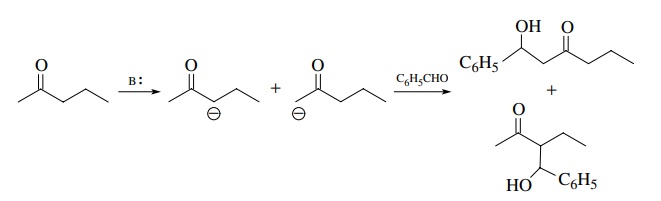
easily converted to their enolates. Ketones, however, are problematic with
regard to the regioselectivity of enolate formation if they are unsymmetric. As
seen in the following example, two regioisomeric enolates can be produced by
the removal of the nonequivalent α
protons of 2-pentanone by base, and thus two regioisomeric aldol products are
possible.
Regiochemical
control of enolate formation is thus an important consideration when planning
ways to construct a carbon–carbon bond using a ketone enolate. There are
several strategies for controlling the regiochemistry of proton removal.
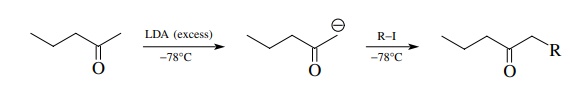
The
first is to take advantage of the fact that the less-substituted α position has slightly more acidic
protons. If a ketone is added slowly
to a cold solution of LDA, the more acidic proton will be removed
preferentially. The resulting enolate is termed the kinetic enolate because the more acidic proton is removed faster than the less acidic proton. Both
steric and electronic factors contribute to the more rapid removal of protons
from the less highly substituted α
carbon.
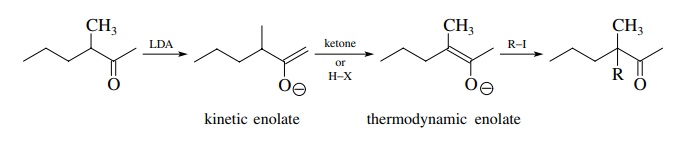
The
enolate that is the most stable usually has the most highly substituted dou-ble
bond and is called the thermodynamic enolate. If a slight excess of the ketone
is used or a trace of protic impurities is present, equilibrium between the
enolates is established and isomerization to the more highly substituted
enolate occurs.
The
thermodynamic enolate is lower in energy so it is the one favored if
equilib-rium is achieved. For this reason, great care must be taken in the
preparation and reaction of the kinetic enolate so that equilibration does not
occur. On the other hand, preparation and reaction of the thermodynamic enolate
is much easier and demands less rigorous reaction conditions.
Besides
the direct formation of kinetic or thermodynamic enolates of ketones, other
strategies can be employed to produce regiospecific products. An older and
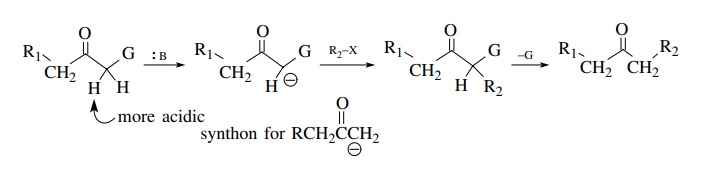
extremely valuable strategy for making the synthetic equivalent of a particular
regiospecific enolate utilizes some group (G) to acidify a proton α to a ketone so that it is removed
preferentially by base. The resulting enolate is used as a carbon nucleophile
and then the group (G) is removed. In this way it appears that one α position of a ketone has been
regioselectively transformed when, in fact, the group G has guided the
chemistry in the reactant but is not present in the product.
The
most common group G is an ester function (although many other groups have been
employed as well). The starting β-ketoester,
which can be prepared easily by a Claisen-type reaction of an ester enolate and
an acid chloride, has a very acidic α
proton (pKa ∼
9–10) which is easily removed (i.e., 1 equiv. NaH or 1 equiv. EtO−).
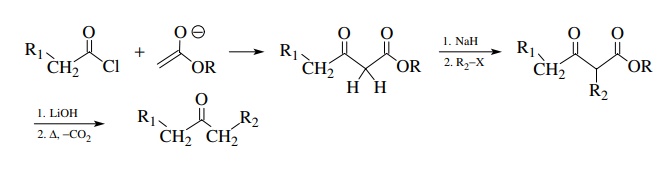
The
resulting enolate is used as a nucleophile to form a new carbon–carbon bond.
The ester is then hydrolyzed and CO2 is thermally ejected to provide
an α-substituted ketone. This
strategy is simple, efficient, and convenient and is widely used. This
synthesis is commonly referred to as the acetoacetic ester synthesis since the
most simple starting material is an ester of acetoacetic acid if R1 = H.
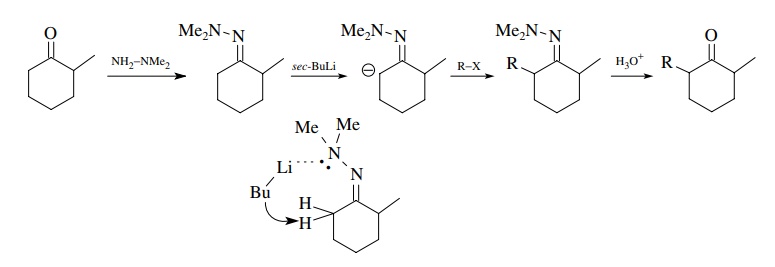
A
third strategy for controlling enolate formation is to convert the carbonyl
group to a N ,N -dimethylhydrazone. The hydrazone is less reactive than the
carbonyl group, and removal of an α
proton by a strong base takes place at the least hindered α position. Alkylation followed by hydrolysis gives back carbonyl
product that is the same as the result of kinetic control of enolate formation.
However, this method does not have the problems of equilibration as found for
simple enolate formation. The regioselectivity of proton removal from the
hydrazone is probably related to the geometry of the hydrazone. The
dimethylamino group is pointed toward the least hindered α position for steric reasons and directs the base to that position
by coordination with the lone pairs on nitrogen.
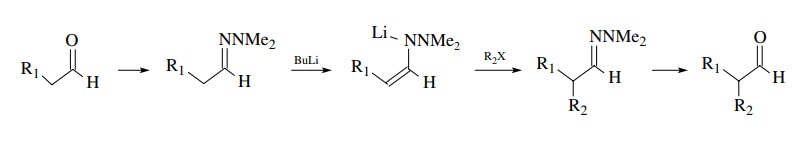
The
use of hydrazones is particularly important to form the enolate equivalents of
aldehydes. Aldehydes are quite reactive as electrophiles, so as soon as some
enolate has been formed, it reacts with the unreacted aldehyde present in
solution. Conversion of the aldehyde to its N
,N -dimethylhydrazone (=NNMe2) lowers the
electrophilicity so that α-proton
removal can take place and then the electrophile of choice can be added.
Hydrolysis gives back the aldehyde. In this case the geometry of the hydrazone
is unimportant since aldehydes have only one α position from which protons can be removed by base.
Related Topics
