Pd(0)-Catalyzed Carbon-Carbon Bond Formation
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Carbon-Carbon Bond Formation Between Carbon Nucleophiles and Carbon Electrophiles
The most important catalytic reactions for the formation of carbon–carbon bonds involve the chemistry of Pd(0).
Pd(0)-CATALYZED CARBON–CARBON
BOND FORMATION
The
most important catalytic reactions for the formation of carbon–carbon bonds
involve the chemistry of Pd(0). Complexes of zero-valent palladium such as
Pd(PPh3)4 are available commercially or can be prepared
in situ by the reaction of Pd(II) salts [e.g., Pd(OAc)2, PdCl2,
etc.] with phosphines or other reductants. The most stable complexes are those
in which the sum of d electrons from the metal and electrons donated by ligands
totals 18. Palladium(0) has a d10 electron configuration and thus
normally coordinates with four ligands which each donate a pair of electrons to
the metal. Such complexes are said to be coordinatively saturated and tend to
be stable and relatively unreactive. However, dissociation of one or two
ligands in solution produces a 16- or 14-electron complex which is reactive and
seeks to regain the 18-electron configuration.
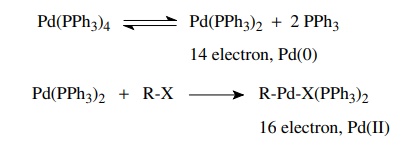
Palladium(0)-catalyzed
transformations generally involve three steps: oxida-tive addition, insertion
or transmetallation (really a special type of insertion), and reductive
elimination. Together they comprise a pathway for the formation of new
carbon–carbon bonds. Oxidative addition takes place when a coordinatively
unsaturated Pd(0) species cleaves a covalent bond to give a new complex in
which the palladium is oxidized to Pd(II). Typically dissociation of two
phos-phine ligands to a 14-electron complex is the first step followed by
oxidative addition to give a 16-electron Pd(II) complex.
Oxidative
addition
This
is quite analogous to the formation of a Grignard reagent by the oxidative
addition of Mg(0) to an alkyl halide. What is remarkable is the generality and
functional tolerance of the palladium process. A variety of bonds undergo
oxida-tive addition with Pd(0). Bonds from carbon to halogen and other good
leaving groups such as sulfonates, esters, and phosphonates are used most often
(and often referred to as C–X or R–X bonds), but many other bond types are
known to react. Even though the product of oxidative insertion has a
carbon–palladium bond, this bond is unaffected by most functional groups. Thus
alcohols, amines, amides, esters, ketones, aldehydes, and even carboxylic acids
can be present in the substrate without interfering with the addition reaction
or subsequent reactions. This is a truly phenomenal tolerance for
functionality!
Another
interesting facet of Pd(0) oxidative insertion is the chemoselectivity of the
process. The most reactive bonds are vinyl and aryl C–X bonds, whereas with
most other metals these are the least reactive bond types. Palladium(0) also
inserts into allylic halides and esters, acid halides, and several other bonds
but reacts only sluggishly with C–X bonds to saturated carbon. Taken together
these characteristics make Pd(0) chemistry nearly unique.
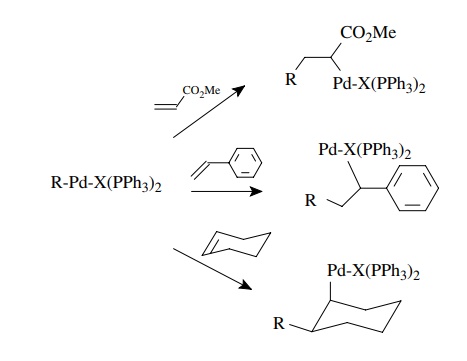
The
second step is insertion or transmetallation. An insertion reaction occurs when
the palladium–carbon bond adds across a π
bond to give a new organopal-ladium species. The types of π bonds normally reactive include alkenes, dienes, alkynes, carbon
monoxide, and sometimes carbonyl π
bonds. By far the most common reactions use alkenes and alkynes for the
insertion reaction. This step results in a new carbon–carbon bond.
Insertion
R-Pd-X(PPh3)2
+ A=B → R-A-B-Pd-X(PPh3)2
for
example,
R-Pd-X(PPh3)2 + CH2 = CH2 → R-CH2-CH2-Pd-X(PPH3)2
The
regiochemistry of the insertion results from a combination of factors which are
still being sorted out. It is possible to think of the carbon attached to
palladium as electron rich, and it tends to attack the π system at the least electron rich position. Thus alkenes with
electron-withdrawing groups react faster than alkenes with electron-donating
groups. It is quite paradoxical, however, that alkenes, dienes, and alkynes
react much more readily than carbonyl compounds, even though the latter are
much more electron deficient.
Moreover
there appears to be a steric bias which causes the R group to attack the least
hindered end of the π system. In
cases where the two ends of the π
bond are similar or where electron-donating groups are attached, the
regiochemistry can be very sensitive to the reaction conditions and the ligands
that coordinate to palladium. At this point controlling the regiochemistry in
such systems is more art than science! Nevertheless, in most cases it is
possible to predict the regiochemistry with good success. Finally the
stereochemistry of the insertion is syn; thus the insertion appears to be a
concerted 1,2 addition across one face of the π system.
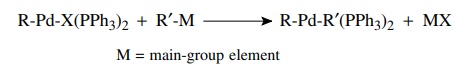
Transmetallation
occurs when compounds with bonds from carbon to several main-group elements
(e.g., B, Al, Sn, Si, Hg) are present in the reaction mixture. The palladium
intermediate from oxidative addition can undergo exchange of palladium and the
main-group element. This essentially yields a second carbon ligand bonded to
palladium. The most common compounds used for transmet-allation are tributyl
tin compounds (R–SnBu3) and boronic acids [R–B(OH)2].
Again the most common and successful examples have the main-group element
bonded to an aromatic ring or an alkene.
Transmetallation
for
example,
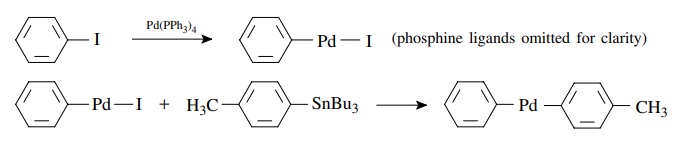
The
last step is reductive elimination in which the organic product is liberated
and Pd(0) is regenerated to begin the catalytic cycle again. When there are two
carbon ligands attached to palladium, as is the case when a transmetallation
has occurred, the two carbon fragments couple with the expulsion of Pd(0). This
occurs rapidly after a transmetallation and in these instances is the step in
which carbon–carbon bond formation occurs.
Reductive
elimination
R-Pd-R′(PPh3)2
→ R-R′ + Pd(PPh3)2
for
example,
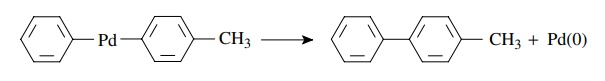
A
second common reductive elimination process termed β-hydride elimination occurs when there is a hydrogen atom β to the carbon–palladium bond, as in
the case where an insertion reaction has taken place. The palladium atom
inserts into the β carbon–hydrogen
bond to give a palladium hydride species coordinated to the alkene. This is a
reversible reaction and is akin to the process of alkene hydrogenation
catalyzed by palladium. Dissociation of the alkene and elimination of HX gives
back the Pd(0) catalyst. Since a strong acid is liberated in the β elimination, a base such as
triethylamine is usually added to the reaction mixture to scavenge this acid.
Although the formation of alkenes by β-hydride
elimination is a facile process, it is not possible to form an alkyne or allene
by β-hydride elimination from a vinyl
palladium species.
β-Hydride elimination
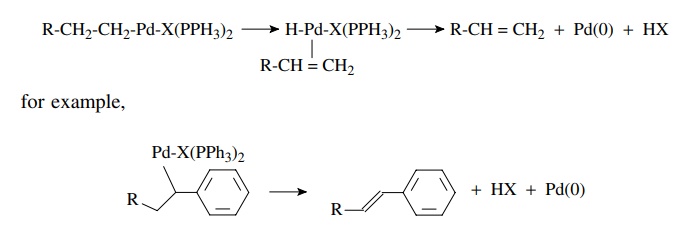
While
the above reactions represent only a small fraction of the reactions known for
palladium, they form the basis of a powerful methodology for building carbon
structures. Several variations have been developed which utilize certain types
of reactants and give particular types of products. All these variations,
however, contain a common theme. In each case an electron-deficient reagent
(e.g., a vinyl halide or aromatic triflate) reacts with an electron-rich
reagent (e.g., an alkene, an organoborane, or an organotin) with the formation
of a new carbon–carbon bond. In that sense these reactions are related to the
reactions between carbon nucleophiles and carbon electrophiles discussed previously
in this chapter. They are quite different, however, because they proceed only in the presence of Pd(0). In fact
they proceed only in the coordination
sphere of Pd(0). The ability of Pd(0) to catalyze these reactions is nearly
unique! We will now examine some of the more common processes.
Heck Reaction
The
Heck reaction involves the coupling of an organopalladium species formed by
oxidative addition to an alkene followed by β-hydride
elimination. The product is an alkene in which a vinyl hydrogen on the original
alkene is replaced by the organic group on palladium. Thus aryl and alkenyl
halides can be coupled to alkenes.
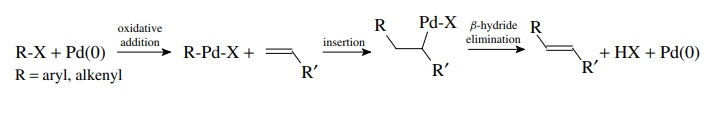
Because
the by-product of the coupling is a strong acid, bases are usually added to the
reaction mixture to scavenge it. For example, 4-iodobromobenzene can be coupled
with methyl acrylate to give the 4-bromocinnamate ester in >68% yield. This reaction takes advantage of the faster oxidative
addition to the carbon–iodine bond to give a single product.
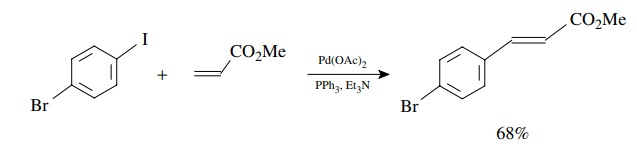
The
Heck reaction was discovered in the early 1970s and is extremely useful for
rapidly assembling carbon skeletons. This reaction is unique to palladium! A
great deal of information is known about the reaction. For example, the success
of the reaction depends on each of the three steps involved. Electron-donating
groups decrease the reactivity of alkenyl halides and triflates toward Pd(0),
whereas electron-withdrawing group increase the rate of oxidative addition. In
cases where Pd(II) salts are used, it is assumed that they are converted to
Pd(0) by some redox process.
The
insertion reaction is stereospecific and syn. Moreover the β-hydride elimi-nation is also syn. For acyclic alkenes there is
free rotation in the organopalladium intermediate so that the more stable trans-alkene is formed.
Electron-withdrawing groups in the alkene also increase the rate of the
insertion reaction and give higher yields generally, but the reaction is
limited to relatively sterically unhin-dered alkenes. In general, polar
solvents such as DMF or acetonitrile are most commonly used. There are several
common additives which aid in the reaction. These include lithium or
tetraalkylammonium chlorides and bromide, silver salts, or cuprous iodide, but
exactly how they function is unknown at present.
The
conversion of carbonyl compounds to their enol triflates provides a very simple
way to couple the carbonyl carbon to an alkene. In general, however, aryl and
vinyl iodides are the preferred substrates because of their ease of oxidative
addition. Terminal alkynes are also good coupling partners.
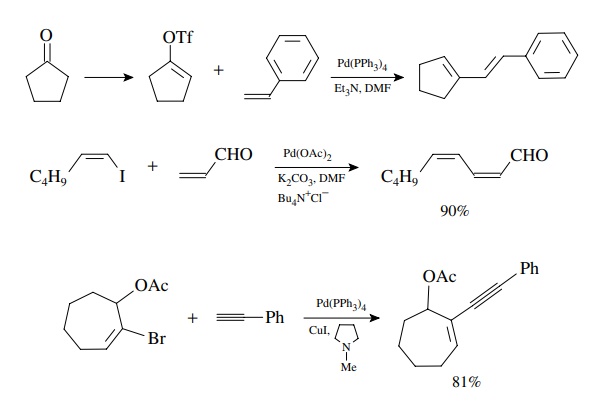
Intramolecular
versions of the Heck reaction are very useful for the construc-tion of ring
systems. The entropic advantage of having both coupling partners present in the
same molecule increases the efficiency of the insertion reaction and leads to
efficient reactions. Moreover the intramolecular version can be carried out on
hindered substituted alkenes, whereas the intermolecular Heck reaction is
largely restricted to monosubstituted alkenes. These reactions illustrate the
syn stereochemistry of both the insertion reaction and the elimination. A number
of multicyclic natural products have been synthesized using intramolecular Heck
reactions to assemble the skeletons, and this has become a powerful synthetic
tool for such compounds.
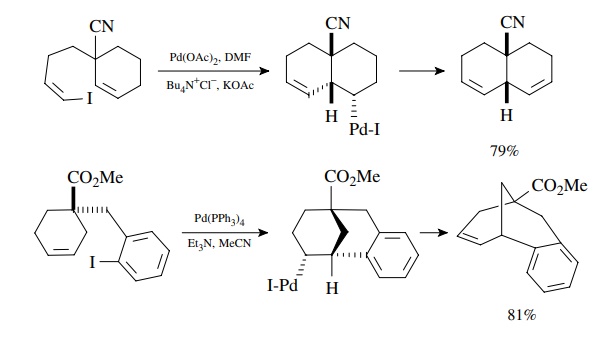
Suzuki Coupling
The
coupling of organoboron compounds with aryl or alkenyl halides is called the
Suzuki reaction and was discovered in the early 1980s. This is a tremendously
versatile method for joining two carbon fragments and is widely used in the
com-mercial manufacture of pharmaceuticals, in the synthesis of compound
libraries, and in drug discovery. After oxidative addition to the halide, the
organopalladium intermediate undergoes transmetallation with the boronic acid
or ester. The new carbon–carbon bond is formed in the reductive elimination
which produces the product and regenerates the Pd(0) catalyst. A base must be
present for the trans-metallation to proceed, and oxybases such as alkoxides,
carbonates, or hydroxide are most commonly employed. The reaction is highly
tolerant of a wide variety of functional groups and thus extremely versatile.
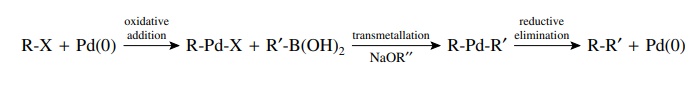
As
noted for the Heck reaction, aryl, alkenyl, and alkynyl bromides, iodides, and
triflates are best for the oxidative addition. However, aromatic,
heteroaro-matic, alkenyl, and even alkyl boronic acids and esters can be
coupled effectively. The reaction appears almost oblivious to other functional
groups present!
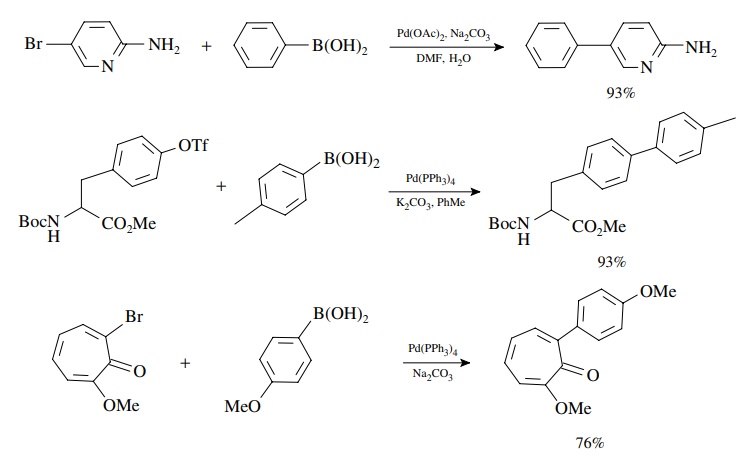
Since
the oxidative addition occurs with retention of configuration and the
transmetallation is also stereospecific with retention, the method is extremely
valuable for the stereoselective synthesis of conjugated dienes. The
stereochemistry of the products is determined by the stereochemistry of the
coupling precursors.
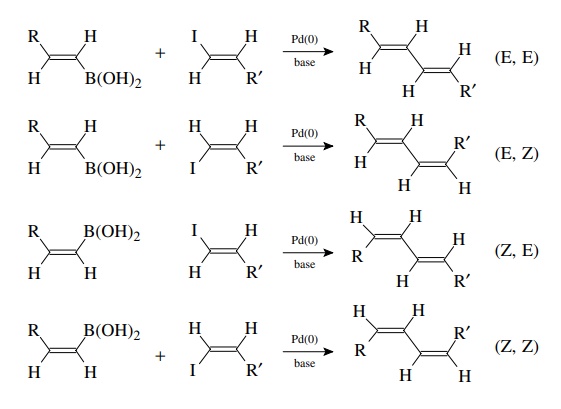
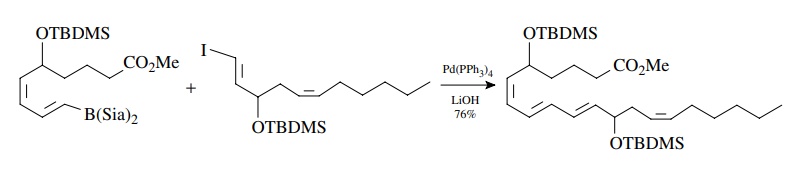
The
required vinyl boranes and vinyl iodides can both be easily made by the
hydroboration of alkynes with disiamyl borane (Sia). Thus the Suzuki reac-tion
is an important methodology for the synthesis of conjugated polyene natu-ral
products.
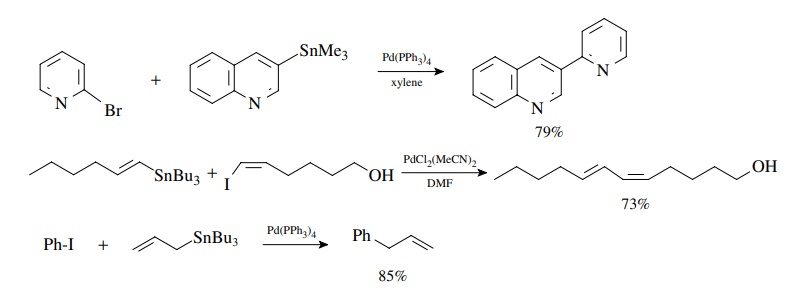
Stille Coupling
Stille
coupling was also developed in the early 1980s and is similar to Suzuki
coupling in its sequence. It is used to couple aryl or vinyl halides or
triflates with organotin compounds via oxidative addition, transmetallation,
and reduc-tive elimination. The oxidative addition reaction has the same
requirements and preferences as discussed earlier for the Heck and Suzuki
reactions. The reduc-tive elimination results in formation of the new
carbon–carbon bond. The main difference is that the transmetallation reaction
uses an organotin compound and occurs readily without the need for an oxygen
base. Aryl, alkenyl, and alkyl stannanes are readily available. Usually only
one of the groups on tin enters into the coupling reaction, and different
groups transfer to palladium with different selectivities. Since simple alkyl
groups have the lowest transfer rate, the most common tin reagents have three
simple alkyl groups (usually methyl or n-butyl).
The fourth group which is transferred is alkynyl, aryl, alkenyl, benzyl, or
allyl.
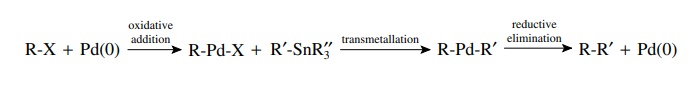
Coupling
of an aryl triflate with an arylstannane is a good method for the preparation
of biaryls and other bis-aromatic species of all types. Coupling of vinyl
groups takes place with retention of stereochemistry. Furthermore transfer of
the allyl group occurs smoothly.
This
is very robust chemistry that works very well with enol triflates.
Intramolecular reactions have been used to close rings of many sizes, including
large rings.
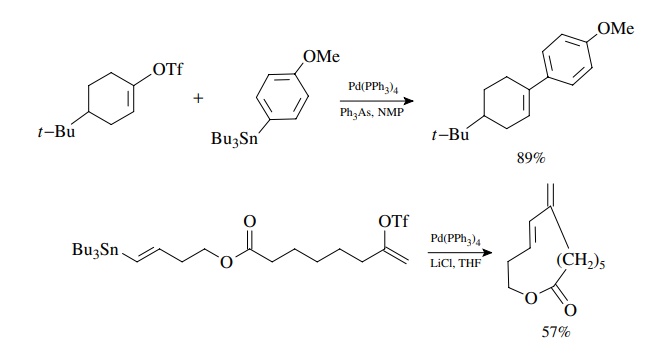
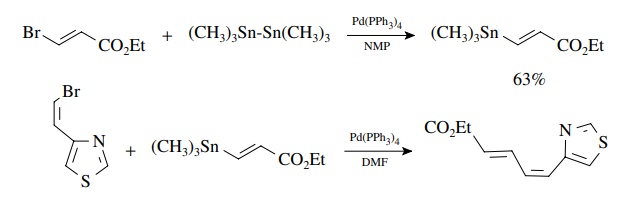
The
use of (Me3Sn)2 provides a unique way to convert vinyl
and aryl halides into the very tin reagents needed for subsequent Stille
couplings!
Related Topics
