Diuretics
| Home | | Pharmacology |Chapter: Essential pharmacology : Diuretics
Based on the diuretic action of calomel, organomercurials given by injection were introduced in the 1920s and dominated for nearly 40 years. The CAse inhibitors were developed in the 1950s from the observation that early sulfonamides caused acidosis and mild diuresis.
DIURETICS
These are drugs which cause a net loss of Na+ and water in urine.
Based on the diuretic
action of calomel, organomercurials given by injection were introduced in the
1920s and dominated for nearly 40 years. The CAse inhibitors were developed in
the 1950s from the observation that early sulfonamides caused acidosis and mild
diuresis. The first modern orally active diuretic chlorothiazide was discovered in 1957, and by early 1960s its
congeners (thiazide diuretics) were already in common use. Availability of furosemide and ethacrynic acid by mid 1960s revolutionized the pattern of diuretic use. The K+ sparing diuretics spironolactone and triamterene were developed in parallel to these.
Diuretics are among the most widely prescribed drugs.
Application of diuretics to the management of hypertension has outstripped their
use in edema. Availability of diuretics has also had a major impact on the
understanding of renal physiology.
Classification
1. High Efficacy
Diuretics (Inhibitors of Na+K+2Cl¯ cotransport)
Sulphamoyl derivatives
Furosemide,
Bumetanide, Torasemide
2. Medium Efficacy Diuretics (Inhibitors of Na+Cl¯ symport)
a.
Benzothiadiazines (Thiazides)
Hydrochlorothiazide, Benzthiazide, Hydroflumethiazide, Clopamide
b.
Thiazide Like (Related Heterocyclics)
Chlorthalidone, Metolazone,
Xipamide, Indapamide.
3. Weak Or
Adjunctive Diuretics
a.
Carbonic Anhydrase Inhibitors
Acetazolamide
b.
Potassium Sparing Diuretics
i.Aldosterone
Antagonist: Spironolactone
ii.Inhibitors
Of Renal Epithelial Na+ Channel: Triamterene, Amiloride.
c. Osmotic
Diuretics
Other high ceiling
diuretics, viz. ethacrynic acid and
organomercurials (mersalyl) are only
historical.
HIGH CEILING (LOOP) DIURETICS
(Inhibitors of Na+K+2Cl¯
Cotransport)
Furosemide (Frusemide) Prototype drug
The development of
this orally and rapidly acting highly efficacious diuretic was a breakthrough.
Its maximal natriuretic effect is much greater than that of other classes. The
diuretic response goes on increasing with increasing dose: upto 10 L of urine
may be produced in a day. It is active even in patients with relatively severe
renal failure. The onset of action is prompt (i.v. 2–5 min., i.m. 10–20 min.,
oral 20–40 min.) and duration short (3–6 hours).
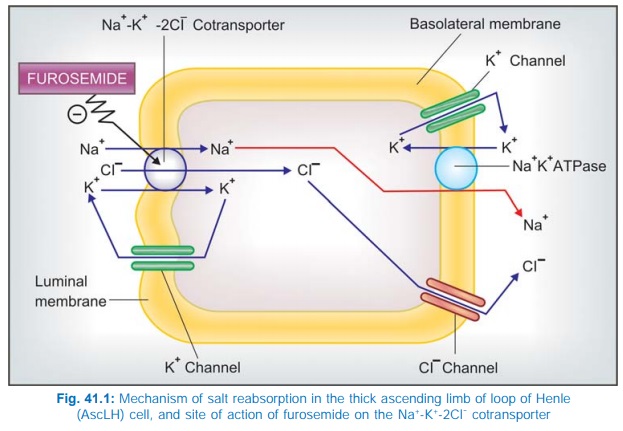
The major site of
action is the thick AscLH (site II) where furosemide inhibits Na+ K+2Cl¯
cotransport (Fig. 41.1). A minor component of action on PT has also been
indicated. It is secreted in PT by organic anion transport and reaches AscLH
where it acts from luminal side of the membrane. It abolishes the corticomedullary
osmotic gradient and blocks positive as well as negative free water clearance.
K+ excretion is increased mainly due to high Na+ load reaching DT. However, at
equinatriuretic doses, K+ loss is less than that with thiazides.
Furosemide has weak
CAse inhibitory action and increase HCO3¯ excretion as well; urinary
pH may rise but the predominant urinary anion is Cl¯; acidosis does not
develop. Its action is independent of acidbase balance of the body and it
causes little distortion of the same; mild alkalosis occurs at high doses.
In addition to its prominent tubular action, furosemide causes
acute changes in renal and systemic haemodynamics. After 5 min of i.v.
injection, renal blood flow is transiently increased and there is redistribution
of blood flow from outer to midcortical zone; g.f.r. generally remains
unaltered due to compensatory mechanisms despite increased renal blood flow.
Pressure relationship between vascular, interstitial and tubular compartments is
altered, the net result of which is decreased PT reabsorption. The intrarenal
haemodynamic changes are brought about by increased local PG synthesis.
Intravenous furosemide
causes prompt increase in systemic venous capacitance and decreases left ventricular
filling pressure, even before the saluretic response is apparent. This action
also appears to be PG mediated and is responsible for the quick relief it
affords in LVF and pulmonary edema.
Furosemide increases
Ca2+ excretion (contrast thiazides which reduce it) as well as Mg2+ excretion.
It tends to raise blood uric acid level by competing with its proximal tubular
secretion as well as by increasing reabsorption in PT which is a consequence of
reduced e.c.f. volume.
The magnitude of hyperuricaemia is lower than that with
thiazides. Hyperglycaemic action of furosemide is also less marked than
thiazides.
Molecular Mechanism Of Action:
A glycoprotein with 12 membrane spanning domains has been
found to function as the Na+K+2Cl¯ cotransporter in many epithelia performing
secretory/ absorbing function, including AscLH. Recently, distinct absorptive or secretory isoforms of Na+K+2Cl¯ cotransporter have been isolated.
The former is exclusively expressed at the luminal membrane of thick
AscLH—furosemide attaches to the Cl¯ binding site of this protein to inhibit
its transport function. The secretory form is expressed on the basolateral
membrane of most glandular and epithelial cells.
Pharmacokinetics
Furosemide is rapidly absorbed orally but bioavailability is
about 60%. In severe CHF oral bioavailability may be markedly reduced. Lipidsolubility
is low, and it is highly bound to plasma proteins. It is partly conjugated with
glucuronic acid and mainly excreted unchanged by glomerular filtration as well
as tubular secretion. Some excretion in bile and directly in intestine also
occurs. Plasma t½ averages 1–2 hour but is prolonged in patients with pulmonary
edema, renal and hepatic insufficiency.
Dose Usually 20–80 mg once daily in the morning. In renal insufficiency, upto 200 mg 6 hourly has
been given by i.m./i.v. route. In pulmonary edema 40–80 mg may be given i.v.
LASIX 40 mg tab., 20 mg/2 ml inj. LASIX HIGH DOSE 500 mg tab,
250 mg/25 ml inj; (solution degrades spontaneously on exposure to light), SALINEX
40 mg tab, FRUSENEX 40, 100 mg tab.
Bumetanide
It is similar to
furosemide in all respects, but is 40
times more potent. It induces very rapid diuresis and is highly effective in
pulmonary edema. However, the site of action, ceiling effect, renal haemodynamic
changes and duration of action are similar to furosemide. A secondary action in
PT has also been demonstrated. It may act in some cases not responding to
furosemide. Hyperuricaemia, K+ loss, glucose intolerance and ototoxicity are
claimed to be less than with furosemide. However, it may rarely cause myopathy.
Bumetanide is more
lipidsoluble, 80–100% bioavailable orally, extensively bound to plasma
proteins, partly metabolized and partly excreted unchanged in urine. Its
accumulation in tubular fluid is less dependent on active secretion. Plasma t½
~60 min, gets prolonged in renal and hepatic insufficiency.
Dose: 1–5 mg oral OD in the morning,
2–4 mg i.m./i.v., (max. 15 mg/day in
renal failure).
BUMET, 1 mg tab., 0.25
mg/ml inj.
Torasemide (Torsemide)
Another high ceiling diuretic with properties similar to
furosemide, but 3 times more potent. Oral absorption is more rapid and more complete.
The elimination t½ (3.5 hours) and duration of action (4–8 hours) are longer.
Torasemide has been used in edema and in hypertension.
Dose: 2.5–5 mg OD in
hypertension; 5–20 mg/day in edema;
upto 100 mg BD in renal failure.
DIURETOR 10, 20 mg
tabs, DYTOR 10, 20, 100 mg tabs.
Use Of High Ceiling Diuretics
a) Edema Diuretics are used
irrespective of etiology of edema—cardiac,
hepatic or renal. The high ceiling diuretics are preferred in CHF for rapid
mobilization of edema fluid (see Ch.
No. 37). Thiazides may be used for maintenance, but often prove ineffective and
high ceiling drugs are called in. For nephrotic and other forms of resistant edema,
the high ceiling diuretics are the drugs of choice. In chronic renal failure
massive doses have to be used, but they continue to be effective while
thiazides just do not produce any action. In impending acute renal failure, loop
diuretics may decrease the need for dialysis.
b) Acute Pulmonary Edema (Acute LVF, Following MI): Intravenous administration of furosemide or its congeners produces prompt relief. This is due to vasodilator action that precedes the saluretic action. Subsequently, decrease in blood volume and venous return is responsible for the improvement.
c) Cerebral Edema: Though osmotic
diuretics are primarily used,
furosemide may be combined to improve efficacy.
d) Hypertension: High ceiling diuretics
are indicated only in presence
of renal insufficiency, CHF, in resistant cases or hypertensive emergencies;
otherwise thiazides are preferred.
e) Along with blood
transfusion in severe anaemia, to prevent vascular overload.
f) Hypercalcaemia and renal
calcium stones: because furosemide and its congeners increase calcium excretion
and urine flow, they may help to reduce serum calcium level. Excess salt that
is lost must be replaced.
Forced diuresis with saline and furosemide infusion is no longer
recommended to treat poisonings.
THIAZIDE AND RELATED DIURETICS
(Inhibitors of Na+Cl¯ symport)
Chlorothiazide was
synthesized as a CAse inhibitor variant which produced urine that was rich in
Cl¯, and diuresis occurred in alkalosis as well as acidosis. A large number of
congeners were developed subsequently and the thiadiazine ring was replaced by
other heterocyclic rings, but the type of activity remained the same. The
important features of agents marketed in India are presented in Table 41.1.
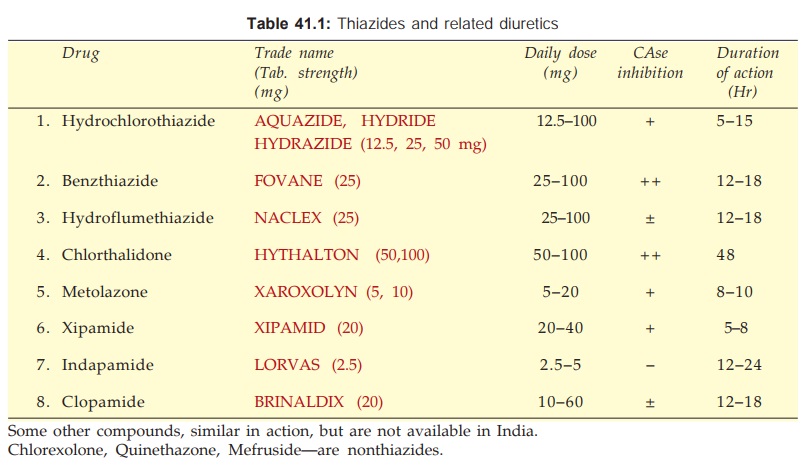
These are medium
efficacy diuretics with primary site of action in the cortical diluting segment
or the early DT (Site III). Here they inhibit Na+–Cl¯ symport at the luminal
membrane. They do not affect the corticomedullary osmotic gradient indicating
lack of action at the medullary thick AscLH. Positive free water clearance is
reduced (very dilute urine cannot be passed in the absence of ADH), but
negative free water clearance (in the presence of ADH) is not affected. This
strengthens the view that the site of action is in between thick AscLH and late
DT. These drugs gain access to their site of action via organic acid secretory pathway in PT and then along the tubular
fluid to early DT, where they bind to specific receptors located on the luminal
membrane. Like the Na+ K+2Cl¯ cotransporter, the Na+Cl¯ symporter is also a
glycoprotein with 12 membrane spanning domains that binds thiazides but not
furosemide or any other class of diuretics. It has been cloned and shown to be
selectively expressed on the luminal membrane in the DT. The site of action of
thiazide diuretics is shown in Fig. 41.2.
Some of the thiazides
and related drugs have additional CAse inhibitory action in PT; intensity of
this action differs among different compounds (Table 41.1), but it is generally
weak and clinically insignificant. However, it may confer some proximal tubular
action to the compounds.
Under their action,
increased amount of Na+ is presented to the distal nephron, more of it
exchanges with K+ → urinary K+ excretion is increased in parallel to the
natriuretic response. The maximal diuresis induced by different agents falls in
a narrow range; though potency (reflected in daily dose) differs markedly.
Nevertheless, they are moderately efficacious diuretics because nearly 90% of
the glomerular filtrate has already been reabsorbed before it reaches their
site of action. They have a flat dose response curve; little additional
diuresis occurs when the dose is increased beyond 100 mg of hydrochlorothiazide
or equivalent. They do not cause significant alteration in acid-base balance of
the body.
By their action to
reduce blood volume, as also intrarenal haemodynamic changes, they tend to
reduce g.f.r. This is one reason why thiazides are not effective in patients
with low g.f.r. They decrease renal Ca2+ excretion and increase Mg2+ excretion by
a direct distal tubular action. They also decrease urate excretion by the same
mechanism as furosemide.
The extrarenal actions of thiazides consist
of a slowly developing fall in BP in hypertensives and elevation of blood sugar
in some patients due to decreased insulin release which probably is a
consequence of hypokalaemia.
Pharmacokinetics
All thiazides and related drugs are well
absorbed orally; are administered only by this route. Their action starts within
1 hour, but the duration varies from 8–48 hours (Table 41.1). The more lipidsoluble
agents have larger volumes of distribution (some are also tissue bound), lower
rates of renal clearance and are longer acting. The protein binding is also variable.
Most of the agents undergo little hepatic metabolism and are excreted as suCh.
No. They are filtered at the glomerulus as well as secreted in the PT by
organic anion transport. Tubular reabsorption depends on lipid solubility: the
more soluble ones are highly reabsorbed—prolonging duration of action.
Chlorthalidone It is a particularly
long acting agent with a t½ 40–50
hours, used exclusively as antihypertensive.
Metolazone In common with loop diuretics, it is able to evoke a clinically useful response
even in severe renal failure (g.f.r. ~15 ml/min), and has marked additive
action when combined with furosemide. An additional proximal tubular action has
been demonstrated; PO4 reabsorption that occurs in PT is inhibited. It
is excreted unchanged in urine.
Xipamide It has a pronounced diuretic action similar to low doses of furosemide. Because of
longer duration of action—hypokalemia is more prominent.
Indapamide It has little diuretic action in the usual doses, probably because it is highly lipidsoluble,
is extensively metabolized and only small quantity of unchanged drug is present
in the tubular fluid. However, it retains antihypertensive action and is used
for that purpose only.
Uses
Edema: Thiazides may be used
for mild-to-moderate cases. For mobilization of edema fluid more efficacious
diuretics are preferred, but thiazides may be considered for maintenance therapy.
They act best in cardiac edema, less effective in hepatic or renal edema. They are
powerless in the presence of renal failure. Cirrhotics often develop
refractoriness to thiazides due to development of secondary hyperaldosteronism.
Hypertension: Thiazides and related
diuretics, especially chlorthalidone are one of the first line drugs (Ch. No.
40).
Diabetes Insipidus: They reduce urine volume (see
Ch. No. 42).
Hypercalciuria with recurrent calcium stones in the kidney. Thiazides act by reducing Ca2+
excretion.
Complications Of High Ceiling And Thiazide Type Diuretic Therapy
Most of the adverse effects of these drugs are related to fluid
and electrolyte changes caused by them. They are remarkably safe in low doses used
over short periods. Many subtle metabolic effects have been reported in their long-term
use as antihypertensives at the relatively higher doses used in the past (see Ch. No. 40).
1. Hypokalaemia: This is the most
significant problem. It is rare at
low doses, but may be of grave consequence when brisk diuresis is induced or on
prolonged therapy, especially if dietary K+ intake is low. Degree of hypokalaemia
appears to be related to the duration of action of the diuretic; longer acting
drugs causing more K+ loss. The usual manifestations are weakness, fatigue,
muscle cramps; cardiac arrhythmias are the serious complications. Hypokalaemia
is less common with standard doses of high ceiling diuretics than with
thiazides, possibly because of shorter duration of action of the former which
permits intermittent operation of compensatory repletion mechanisms. It can be
prevented and treated by:
(a) High dietary K+
intake or
(b) Supplements of KCl
(24–72 mEq/day) or
(c) Concurrent use of
K+ sparing diuretics.
Measures (b) and (c)
are not routinely indicated, but only when hypokalaemia has been documented or
in special risk situations, e.g. cirrhotics, cardiac patients—especially post
MI, those receiving digitalis, antiarrhythmics, or tricyclic antidepressants
and elderly patients.
Serum K+ levels are
only a rough guide to K+ depletion, because K+ is primarily an intracellular
ion. Nevertheless, an attempt to maintain serum K+ at or above 3.5 mEq/L should
be made.
Combined tablets of diuretics and KCl are not recommended
because:
·
they generally contain insufficient quantity
of K+ (8–12 mEq only).
·
may cause gut ulceration by releasing KCl at
one spot.
·
K+ is retained better if given
after the diuresis is over.
K+ sparing diuretics
are more efficacious and more convenient in correcting hypokalaemia than are K+
supplements. ACE inhibitors/AT1 antagonists given with thiazides
tend to prevent development of hypokalaemia.
Alkalosis may occur with hypokalaemia, because more H+ exchanges
with Na+ in DT when less K+ is available for exchange.
2. Acute Saline Depletion: Over enthusiastic use of diuretics,
particularly high ceiling ones, may cause dehydration and fall in BP
(especially in erect posture). Serum Na+ and Cl¯ levels remain normal because
isotonic saline is lost. It should be treated by saline infusion.
3. Dilutional
Hyponatraemia: Occurs in CHF patients when vigorous diuresis is induced with
high ceiling agents, rarely with thiazides. Kidney tends to retain water,
though it is unable to retain salt due to the diuretic; e.c.f. gets diluted,
hyponatraemia occurs and edema persists despite natriuresis. Patients feel very
thirsty. Treatment of this distortion of fluidelectrolyte balance is difficult:
withhold diuretics, restrict water intake and give glucocorticoid which
enhances excretion of water load. If hypokalaemia is present, its correction helps.
4. GIT and CNS
Disturbances: Nausea, vomiting and diarrhoea may occur with
any diuretic. Headache, giddiness, weakness, paresthesias, impotence are
occasional complaints with thiazides as well as loop diuretics.
5. Hearing
Loss: Occurs rarely, only
with high ceiling diuretics and when
these drugs are used in the presence of renal insufficiency. Increased
salt content of
endolymph and a direct toxic action on the hair cells in internal ear appear to
be causative.
6. Allergic
Manifestations: Rashes, photosensitivity
occur, especially in patients hypersensitive to sulfonamides. Blood dyscrasias
are rare; any diuretic may be causative.
7. Hyperuricaemia: Long-term use of higher dose thiazides in hypertension has caused rise
in blood urate level. This is uncommon now due to use of lower doses (see Ch. No. 40). Furosemide produces a
lower incidence of hyperuricaemia. This effect can be counteracted by
allopurinol. Probenecid is better avoided, because it may interfere with the
diuretic response, particularly of loop diuretics.
8. Hyperglycaemia
and Hyperlipidemia: Have occurred in the use of diuretics as antihypertensive
(see Ch. No. 40). These metabolic
changes are minimal at low doses now recommended.
9. Hypercalcaemia: Occurs with thiazides, while hypocalcaemia occurs with high
ceiling diuretics when these are
administered chronically.
10. Magnesium Depletion: It may develop after prolonged use of
thiazides as well as loop diuretics, and may increase the risk of ventricular
arrhythmias, especially after MI or when patients are digitalized. K+ sparing
diuretics given concurrently minimise Mg2+ loss.
11. Thiazides have
sometimes aggravated renal insufficiency, probably by reducing
g.f.r.
12. Brisk diuresis
induced in cirrhotics may precipitate mental
disturbances and hepatic coma. It may be due to hypokalaemia, alkalosis and
increased blood NH3 levels.
13. Diuretics should
not be used in toxaemia of pregnancy in which blood volume is low
despite edema. Diuretics may further
compromise placental circulation → miscarriage, foetal death. Thus, diuretics
are contraindicated in pregnancy induced hypertension.
Interactions
1. Thiazides and high
ceiling diuretics potentiate all other antihypertensives. This interaction is
intentionally employed in therapeutics.
2. Hypokalaemia induced
by these diuretics:
·
Enhances digitalis toxicity.
·
Increases the incidence of polymorphic
ventricular tachycardia due to quinidine and other antiarrhythmics.
·
Potentiates competitive neuromuscular blockers
and reduces sulfonylurea action.
3. High ceiling
diuretics and aminoglycoside antibiotics are both ototoxic and nephrotoxic;
produce additive toxicity; should be used together cautiously.
4. Cotrimoxazole given
with diuretics has caused higher incidence of thrombocytopenia.
5. Indomethacin and other
NSAIDs diminish the action of high ceiling diuretics. Inhibition of PG
synthesis in the kidney, through which furosemide and related drugs induce
intrarenal haemodynamic changes which secondarily affect salt output, appears
to be the mechanism. Antihypertensive action of thiazides and furosemide is
also diminished by NSAIDs.
6. Probenecid
competitively inhibits tubular secretion of furosemide and thiazides: decreases
their action by reducing the concentration in the tubular fluid, while
diuretics diminish uricosuric action of probenecid.
7. Serum lithium level
rises when diuretic therapy is instituted. This is due to enhanced reabsorption
of Li+ (and Na+) in PT.
Resistance To High Ceiling Diuretics
Refractoriness
(progressive edema despite escalating diuretic therapy) is more common with
thiazides, but occurs under certain circumstances with high ceiling diuretics
as well. The causes and mechanism of such resistance include:

Long-term use of loop
diuretics causes distal nephron hypertrophy → resistance. Addition
of metolazone, or to some extent a thiazide, which act on distal tubule
overcome the refractoriness in many cases. Further increase in dose and/or
fractionation of daily dose may restart diuresis. Bedrest may also help.
CARBONIC ANHYDRASE INHIBITORS
Carbonic anhydrase (CAse) is an
enzyme which catalyses the reversible reaction H2O + CO2
←→ H2CO3. Carbonic acid spontaneously ionizes H2CO3←→H+
+HCO¯3 (Fig. IX.2).
Carbonic anhydrase thus functions in CO2
and HCO3¯ transport and in H+ ion secretion. The enzyme is present
in renal tubular cell (especially PT) gastric mucosa, exocrine pancreas,
ciliary body of eye, brain and RBC. In these tissues a gross excess of CAse is
present, more than 99% inhibition is required to produce effects.
Acetazolamide
It is a sulfonamide
derivative which noncompetitively but reversibly inhibits CAse in PT cells
resulting in slowing of hydration of CO2 → decreased
availability of H+ to exchange with luminal Na+ through the Na+H+ antiporter.
Inhibition of brush border CAse retards dehydration of H2CO3
in the tubular fluid so that less CO2 diffuses back into the cells.
The net effect is inhibition of HCO¯ (and accompanying Na+) reabsorption in PT → prompt but mild
alkaline diuresis ensues.
Secretion of H+
in DT and CD is also inhibited. Though H+ is secreted at this site
by a H+ATPase, it is generated in the cell by CAse mediated
reaction. As such, this is a subsidiary site of action of CAse inhibitors. When
CAse inhibitors are given, the distal Na+ exchange takes place only
with K+ which is lost in excess. For the same degree of natriuresis
CAse inhibitors cause the most marked kaliuresis compared to other diuretics.
The urine produced under acetazolamide action is alkaline and rich in HCO¯
which is matched by both Na+ and K+. Continued action of
acetazolamide depletes body HCO3¯ and causes acidosis; less HCO3¯
(on which its diuretic action depends) is filtered at the glomerulus → selflimiting diuretic
action. The extrarenal actions of acetazolamide are:
·
Lowering of intraocular tension due to
decreased formation of aqueous humour (it is rich in HCO¯)3.
·
Decreased gastric HCl and pancreatic NaHCO3
secretion: This action requires very high doses—clinically not significant.
·
Raised level of CO2 in brain and
lowering of pH → sedation and elevation of seizure threshold.
·
Alteration of CO2 transport in
lungs and tissues: these actions are masked by compensatory mechanisms.
Pharmacokinetics
Acetazolamide is well
absorbed orally and excreted unchanged in urine. Action of a single dose lasts
8–12 hours.
Uses
Because of selflimiting action, production of acidosis and hypokalaemia, acetazolamide is
not used as diuretic. Its current clinical uses are:
1. Glaucoma: as adjuvant to other ocular hypotensives (see Ch. No. 10).
2. To alkalinise urine: for urinary tract infection or to
promote excretion of certain acidic drugs.
3. Epilepsy: as adjuvant in absence seizures when primary drugs
are not fully effective.
4. Acute mountain
sickness: for symptomatic relief as well as prophylaxis. Benefit occurs
probably due to reduced CSF formation as well as lowering of CSF and brain pH.
5. Periodic paralysis.
Dose: 250 mg OD–BD; DIAMOX, SYNOMAX 250 mg tab. IOPARSR 250 mg SR cap.
Adverse Effects are
frequent.
Acidosis,
hypokalaemia, drowsiness, paresthesias, fatigue, abdominal discomfort. Hypersensitivity
reactions—fever, rashes. Bone marrow depression is rare but serious. It is
contraindicated in liver disease: may precipitate hepatic coma by interfering
with urinary elimination of NH3 (due to alkaline urine). Acidosis is
more likely to occur in patients of COPD.
Some topical CAse
inhibitors have been introduced for use in glaucoma (see Ch. No. 10).
POTASSIUM SPARING DIURETICS
These are either
aldosterone antagonist or directly inhibit Na+ channels in DT and CD cells to
indirectly conserve K+.
Spironolactone (Aldosterone
antagonist)
It is a steroid,
chemically related to the mineralocorticoid aldosterone. Aldosterone acts on
the late DT and CD cells (Fig. 41.3) by combining with an intracellular
mineralocorticoid receptor → induces the formation of ‘aldosterone-induced
proteins’ (AIPs) which promote Na+ reabsorption by a number of mechanisms (see legend to Fig. 41.3) and K+
secretion. Spironolactone acts from the interstitial side of the tubular cell,
combines with the mineralocorticoid receptor and inhibits the formation of AIPs
in a competitive manner. It has no effect on Na+ and K+ transport in the
absence of aldosterone, while under normal circumstances, it increases Na+ and
decreases K+ excretion.
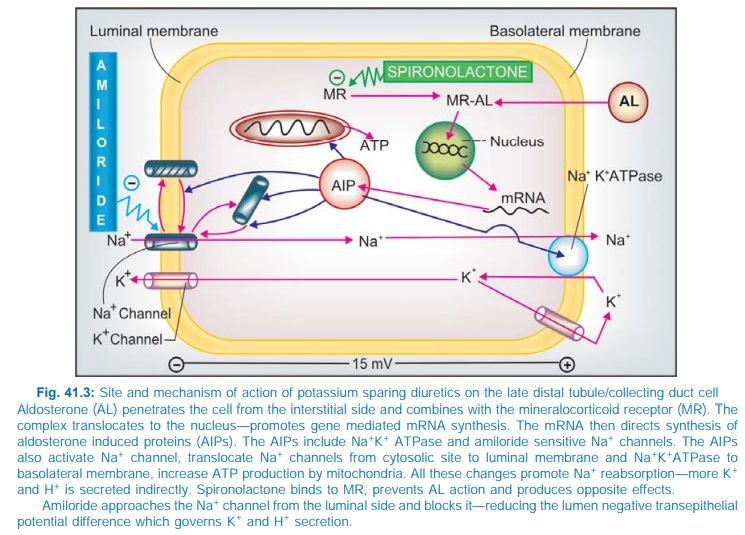
Spironolactone is a
mild saluretic because majority of Na+ has already been reabsorbed proximal to
its site of action. However, it antagonises K+ loss induced by other diuretics
and slightly adds to their natriuretic effect. The K+ retaining action develops
over 3–4 days. It increases Ca2+ excretion by a direct action on renal tubules.
Pharmacokinetics
The oral
bioavailability of spironolactone from
microfine powder tablet is 75%. It is highly bound to plasma proteins and
completely metabolized in liver; converted to active metabolites, the most
important of which is Canrenone that
is responsible for 1/2–2/3 of its action in
vivo. The t½ of spironolactone is 1–2 hours, while that canrenone is ~18
hours. It undergoes some enterohepatic circulation.
Dose: 25–50 mg BD–QID; ALDACTONE 25, 100 mg
tabs.
ALDACTIDE:
Spironolactone 25 mg + hydroflumethiazide 25 mg tab. LACILACTONE, SPIROMIDE:
Spironolactone 50 mg + furosemide 20 mg tab.
Use
Spironolactone is a
weak diuretic in its own right and is used
only in combination with other more efficacious diuretics.
·
Edema: It is more useful in cirrhotic and nephrotic
edema: aldosterone levels are generally high. It breaks the resistance to
thiazide diuretics that develops due to secondary hyperaldosteronism and
reestablishes the response. Thus, it is particularly employed in refractory
edema.
·
To counteract K+ loss due to thiazide and loop
diuretics.
·
Hypertension: Used only as adjuvant to
thiazide to prevent hypokalaemia; has weak antihypertensive action of its own.
·
CHF: As additional drug to conventional
therapy in moderate to severe CHF; can retard disease progression and lower
mortality.
Interactions
·
Given together with K+ supplements— dangerous
hyperkalaemia can occur.
·
Aspirin blocks spironolactone action by
inhibiting tubular secretion of canrenone.
·
More pronounced hyperkalaemia can occur in patients
receiving ACE inhibitors/angiotensin receptor blockers (ARBs).
·
Spironolactone increases plasma digoxin
concentration.
Adverse Effects
The side effects are
drowsiness, confusion, abdominal
upset, hirsutism, gynaecomastia, impotence and menstrual irregularities. Most
serious is hyperkalaemia that may occur especially if renal function is
inadequate. Acidosis is a risk, particularly in cirrhotics. Peptic ulcer may be
aggravated.
Eplerenone is a recently
developed more selective aldosterone antagonist,
that is less likely to cause hormonal disturbances like gynaecomastia,
impotence and menstrual irregularities.
Inhibitors Of Renal Epithelial Na+ Channel
Triamterene and
amiloride are two nonsteroidal organic bases with identical actions. Their most
important effect is to decrease K+ excretion, particularly when it is high due
to large K+ intake or use of a diuretic that enhances K+
loss. This is accompanied by a small increase in Na+ excretion. The
excess urinary Na+ is matched by Cl¯ and variable amounts of HCO3¯
; urine is slightly alkalinized. The effect on urinary electrolyte pattern is
superficially similar to spironolactone, but their action is independent of
aldosterone.
Mechanism Of Action
The luminal membrane
of late DT and CD cells expresses
a distinct ‘amiloride sensitive’ or
‘renal epithelial’ Na+
channel through which Na+ enters the cell down its
electrochemical gradient which is generated by Na+K+
ATPase operating at the basolateral membrane (Fig. 41.3). This Na+
entry partially depolarizes the luminal membrane creating a –15 mV
transepithelial potential difference which promotes secretion of K+ into the
lumen through K+ channels. Though there is no direct coupling between Na+ and
K+ channels, more the delivery of Na+ to the distal nephron—greater is its
entry through the Na+ channel—luminal membrane is depolarized more—driving
force for K+ secretion is augmented. As such, all diuretics acting proximally
(loop diuretics, thiazides, CAse inhibitors) promote K+ secretion. Amiloride and
triamterene block the luminal Na+ channels— indirectly inhibit K+ excretion,
while the net excess loss of Na+ is minor (most of it has already been
reabsorbed).
The intercalated cells
in CD possess an ATP driven H+ pump which secretes H+ ions into the lumen. This
pump is facilitated by lumen negative potential. Amiloride, by reducing the
lumen negative potential, decreases H+ ion secretion as well; predisposes to
acidosis.
Both triamterene and
amiloride are used in conjunction with thiazide type or high ceiling diuretics:
prevent hypokalaemia and slightly augment the natriuretic and antihypertensive
response. Risk of hyperkalaemia is the most important adverse effect of
amiloride and triamterene. These drugs should not be given with K+ supplements;
dangerous hyperkalaemia may develop. Hyperkalaemia is also more likely in
patients receiving ACE inhibitors/ARBs, β blockers, NSAIDs and in those with
renal impairment.
Both drugs elevate
plasma digoxin levels.
Triamterene
It is incompletely
absorbed orally, partly bound to plasma
proteins, largely metabolized in liver to an active metabolite and excreted in
urine. Plasma t½ is 4 hours, effect of a single dose lasts 6–8 hours.
Side Effects are infrequent: consist of nausea, dizziness, muscle cramps and rise in blood
urea. Impaired glucose tolerance and photosensitivity are reported, but urate
level is not increased.
Dose: 50–100 mg daily; DITIDE, triamterene 50
mg + benzthiazide 25 mg
tab;
FRUSEMENE, triamterene
50 mg + furosemide 20 mg tab.
Amiloride
It is 10 times more potent
than triamterene (dose 5–10
mg OD–BD). At higher doses it also inhibits Na+ reabsorption in PT, but this is
clinically insignificant. It decreases Ca2+ excretion and increases urate
excretion. Thus, hypercalcaemic action of thiazides is augmented but
hyperuricaemic action is partly annuled. A mild antihypertensive action is also
reported.
Only ¼ of an oral dose is absorbed. It is not bound to plasma
proteins and not metabolized. The t½ (10–20 hours) and duration of action are
longer than triamterene.
BIDURET, KSPAR: Amiloride 5 mg + hydrochlorothiazide 50 mg tab,
LASIRIDE, AMIMIDE amiloride 5 mg + furosemide 40 mg tab.
Usual side effects are nausea, diarrhoea and headache.
Amiloride blocks entry of Li+ through Na+ channels in the CD cells
and mitigates diabetes insipidus induced by lithium.
Given as an aerosol it affords symptomatic improvement in cystic
fibrosis by increasing fluidity of respiratory secretions.
OSMOTIC DIURETICS
Mannitol
Mannitol is a nonelectrolyte of low molecular weight (182) that
is pharmacologically inert— can be given in large quantities sufficient to
raise osmolarity of plasma and tubular fluid. It is not metabolized in the
body; freely filtered at the glomerulus and undergoes limited reabsorption:
therefore excellently suited to be used as osmotic diuretic. Mannitol appears
to limit tubular water and electrolyte reabsorption in a variety of ways:
·
Retains water iso-osmotically in PT— dilutes
luminal fluid which opposes NaCl reabsorption.
· Inhibits transport processes in the thick
AscLH by an unknown mechanism. Quantitatively this appears to be the most
important cause of diuresis.
· Expands extracellular fluid volume (because it
does not enter cells, mannitol draws water from the intracellular
compartment)—increases g.f.r. and inhibits renin release.
·
Increases renal blood flow, especially to the
medulla—medullary hypertonicity is reduced—corticomedullary osmotic gradient is
dissipated—passive salt reabsorption is reduced.
Though the primary
action of mannitol is to increase urinary volume, excretion of all cations and
anions is also enhanced.
Administration Mannitol is not
absorbed orally; has to be given i.v.
as 10–20% solution. It is excreted with a t½ of 0.5–1.5 hour.
MANNITOL 10%, 20%, in
100, 350 and 500 ml vac.
Uses
Mannitol is never used
for the treatment of chronic edema or as
a natriuretic. Its indications are:
·
Increased intracranial or intraocular tension
(acute congestive glaucoma, head injury, stroke, etc.): by osmotic action it
encourages movement of water from brain parenchyma, CSF and aqueous humour;
1–1.5 g/kg is infused over 1 hour as 20% solution to transiently raise plasma
osmolarity. It is also used before and after ocular/brain surgery to prevent
acute rise in intraocular/intracranial pressure.
·
To maintain g.f.r. and urine flow in impending
acute renal failure, e.g. in shock, severe trauma, cardiac surgery, haemolytic
reactions: 500–1000 ml of the solution may be infused over 24 hours. However,
prognostic benefits in conditions other than cardiac surgery are still
unproven. If acute renal failure has already set in, kidney is incapable of
forming urine even after an osmotic load; mannitol is contraindicated: it will
then expand plasma volume → pulmonary edema and heart failure may develop.
·
To counteract low osmolality of plasma/e.c.f.
due to rapid haemodialysis or peritoneal dialysis (dialysis disequilibrium).
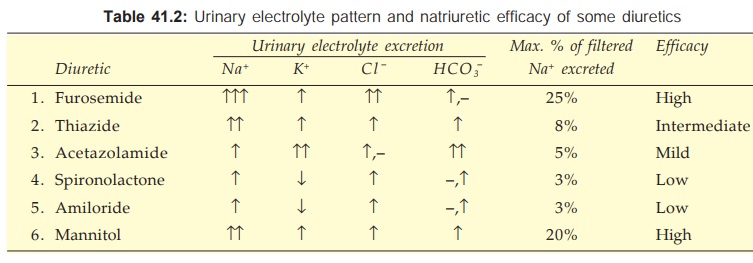
Mannitol along with
large volumes of saline was infused i.v. to produce ‘forced diuresis’ in acute poisonings in the hope of enhancing
excretion of the poison. However, this has been found to be ineffective and to
produce electrolyte imbalances. Not recommended now.
Mannitol is contraindicated in acute tubular
necrosis, anuria, pulmonary edema; acute left ventricular failure, CHF,
cerebral haemorrhage.
Headache due to
hyponatraemia is common, nausea and vomiting may occur; hypersensitivity
reactions are rare.
Isosorbide and Glycerol
These are orally
active osmotic diuretics which may be
used to reduce intraocular or intracranial tension. Intravenous glycerol can
cause haemolysis.
Dose: 0.5–1.5 g/kg as oral
solution.
Related Topics
