Drug development
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Drug development
The process of discovery and development of safe and effective new medicines is long, difficult, and expensive.
Drug development
Introduction
The
process of discovery and development of safe and effective new medicines is
long, difficult, and expensive. A new molecular entity (NME), sometimes also
called a new chemical entity (NCE), is characterized for its potential
therapeutic applications and toxicological profile in nonprimate species, which
is followed by extensive animal and human testing. On aver-age, it costs a
company more than $1 billion and 10–15 years to get one drug from the
laboratory to patients. Only five in ~5000 compounds that enter preclinical
testing make it to human testing. Only one of those five drugs entering human
clinical trials is approved for commercialization.
New
drugs include prescription drugs, over-the-counter (OTC) medications, generic
drugs, biotechnology products, veterinary products, and/or medical devices.
Over-the-counter drugs do not require a physician’s prescription.
Drug
development also focuses on new dosage forms, routes of admin-istration, and
delivery devices for existing drugs. A typical drug discov-ery process entails
target identification, such as a protein or an enzyme whose inhibition may help
in a disease state. The structural features nec-essary in a potential drug
candidate are identified using in silico
molec-ular modeling. Several drugs may be synthesized using combinatorial
chemistry and screened for in vitro
activity in high-throughput assays. The lead candidates are then synthesized in
larger quantities, screened for biological activity, and further optimized to
maximize the affinity, specificity, and potency. A highly specific compound
that only binds the target site is likely to have minimal nontarget effects,
which often lead to adverse effects and toxicity related to the mechanism of
drug action. High affinity for the target site, often resulting in high potency
(low dose for the desired pharmacological effect), minimizes the required dose
of a compound, which can reduce adverse effects and toxicities not asso-ciated
with the drug’s mechanism of action. Such a drug candidate is then identified
as an NME (Figure 2.1), which enters the
development pipeline.
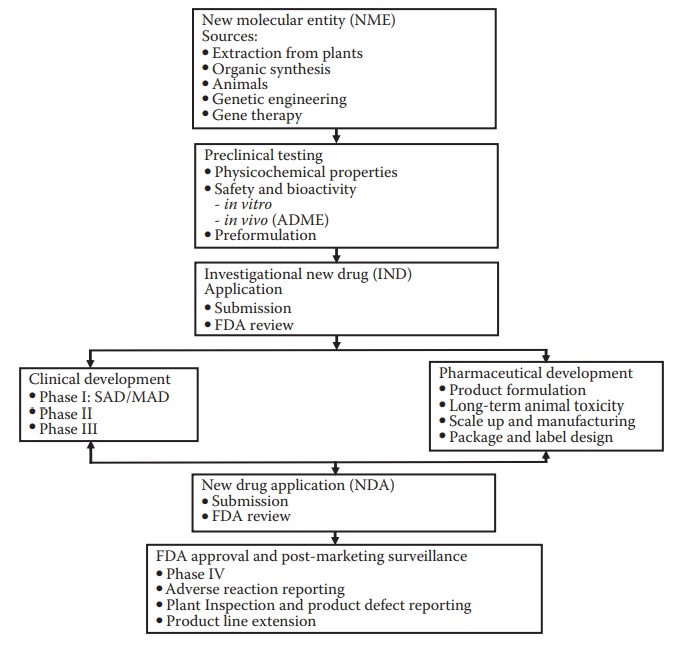
Figure 2.1 Stages of drug development. Drug discovery efforts lead to a new molecu-lar entity (NME) that is identified for development. The NME enters pre-clinical testing and development, which include efficacy and safety testing in animal species. This is followed by the submission of an investigational new drug (IND) application to the regulatory agency, such as the U.S. Food and Drug Administration (FDA). The drug then enters clinical trials: phase I, phase II, and phase III. Pharmaceutical development proceeds concurrent with clinical development and with the objectives of supporting the ongo-ing clinical studies (providing information, documentation, and the drug product for administration to the subjects) and preparation for commer-cialization of the product. Following successful clinical and pharmaceutical development, a data package is submitted to the FDA for approval; this is called the new drug application (NDA). Approval of the NDA is required for the commercialization of a new drug product. Several activities on the drug product continue after commercialization, such as adverse event monitoring, development of line extension products, and additional clinical trials to support label claims or expand target patient populations.
Drug
development studies include preclinical studies, whereby a com-pound is
thoroughly characterized for physicochemical characteristics and is tested in
animal models for toxicity and activity. This is followed by first-in-human
(FIH) phase I studies that test the safety of a compound through sequential
dose-escalating studies, which could be single-dose (called sin-gle ascending
dose, SAD) and/or multiple-dose (called multiple ascending dose, MAD) studies.
In these studies, a subject is administered a SAD or MAD at predefined
intervals, with each successive cohort receiving doses higher than the previous
cohort (hence the term ascending dose studies). Increasingly extensive and
thorough toxicological, pharmacological, and pharmacokinetic characterization
is carried out in humans during phase II and phase III clinical trials (Figure 2.2).
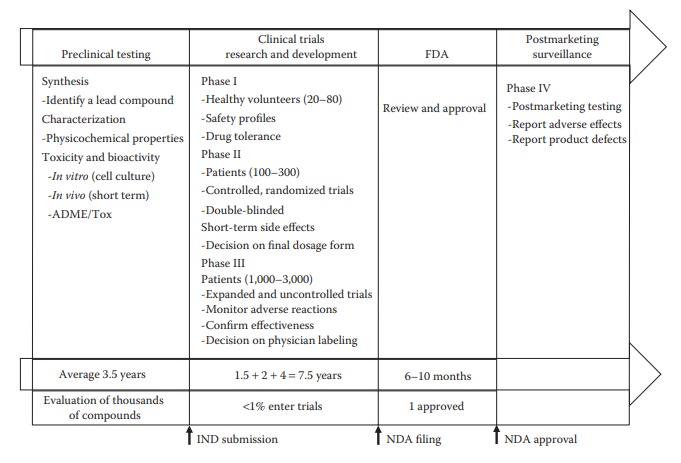
Figure 2.2 Timeline of different phases of drug development. Discovery and preclini-cal testing to identify a lead compound and its detailed characterization for toxicity and bioactivity in vitro and in vivo can take a few years, such as about 3–4 years. A significant amount of time is taken by different phases of clinical trials, about 7–8 years, with increasing duration of time and num-ber of patients required for higher stages of clinical trials. Agency review of the submitted new drug application (NDA) can take several more months, depending on prioritization and workload considerations. A typical medium-to large -sized biopharmaceutical company has several pipeline candidates that are at various stages of development.
Stages
of drug development that precede human testing are termed pre-clinical development, while human testing stage of a drug is
termed clinical development. The
transition from preclinical development to clinical devel-opment requires
regulatory approval through an investigational new drug (IND) application. In
the United States, the sponsor of an NME files an IND with the Food and Drug
Administration (FDA) with preclinical data and proposed protocol for clinical
testing. Unless the FDA has a question or objection to the proposal within 30
days of filing, the drug compound enters FIH (phase I) clinical studies.
Clinical evidence of safety and efficacy of drug products forms the corner-stone of regulatory approval of any new drug product. The clinical evidence is typically gathered in a phased manner, wherein the toxicity or adverse events of a drug are assessed in increasing number of subjects as a molecule progresses through the development timeline (Figure 2.3).
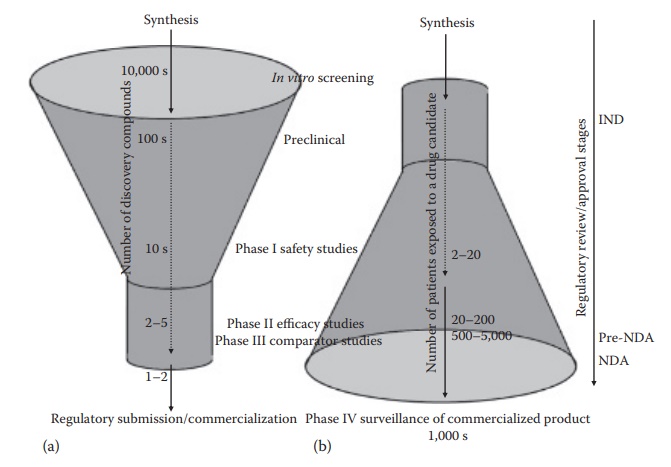
Figure 2.3 Number of compounds proceeding through various stage of drug development— an upright funnel (a), and the number of patients exposed to a given drug through its development—an inverse funnel (b). This figure also illustrates the steps where regulatory review and/or approval are required in the United States, such as the investigational new drug (IND) application submission before initiating phase I studies, a pre-NDA meeting with the FDA after the phase II studies, and new drug application (NDA) submission for drug approval for marketing after the completion of phase III studies. (With kind permission from Springer Science+Business Media: Pharmaceutical Perspectives of Cancer Therapeutics, Anticancer drug development, 2009, 49–92, Narang A.S. and Desai, D.S.)
Pharmaceutical development follows a parallel track with preclinical and clinical development. Pharmaceutical development is responsible for chemistry, manufacturing, and control of both the drug substance and the drug product throughout the life cycle of a compound. This function provides a robust dosage form that meets three key requirements of a drug product: (a) stability, (b) bioavailability, and (c) manufacturability. The key roles of pharmaceutical development are to provide a suitable drug product in a stage-appropriate manner while also ensuring path to future development and commercialization and to bridge the drug product used during different stages of development.
Related Topics
