Collaborating disciplines
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Drug discovery
Various scientific disciplines that work within this paradigm are briefly discussed in the following subsections.
Collaborating
disciplines
New
drug discovery and development is a long, complex process that involves
multidisciplinary scientists working together in diverse, intercon-nected, and
interdependent teams that coordinate their activities and where decision by
each team potentially affects every other team working on a given project. A
consistent feature of new drug discovery research is the cross-disciplinary
progress and collaboration. This interdisciplinary col-laboration is partly
accomplished by scientists working together in different departments within a
pharmaceutical or a biopharmaceutical company. Each of these disciplines is
responsible for one or more aspects of the drug discov-ery and development
process as it moves along the pipeline. These functions are usually known by
different names in different organizations but have common underlying themes,
such as discovery chemistry, discovery biology, preclinical development,
toxicology, pharmaceutical development, clinical development, analytical and
bioanalytical sciences, regulatory sciences, man-ufacturing operations, quality
operations, and commercialization functions.
This
chapter will briefly highlight the roles and responsibilities of each of these
functions, the key methodologies that are adopted in fulfilling those
responsibilities, and the underlying scientific disciplines of study that
con-tribute to drug discovery.
Various
scientific disciplines that work within this paradigm are briefly discussed in
the following subsections.
Biology
The
scientists in this function are responsible for the identification of drug
targets for chosen disease indications and assessment of efficacy of com-pounds
against those targets. The efficacy may be tested in assays that may be
conducted in vitro, ex vivo, or in vivo. These assays, for example, could be target-binding assays
for isolated receptors, cellular response assays in cell culture models, or
animal studies. Dose–response curves are gener-ated in cell culture models (Figure 1.3) that help rank different compounds
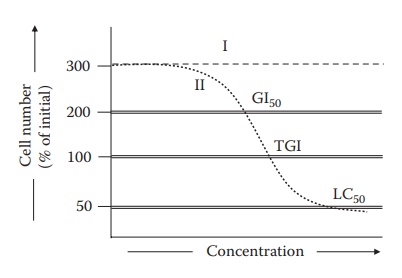
Figure 1.3 An illustration of the
dose–response curves generated during cell culture prescreening. This example
illustrates the dose-dependent cytotoxic effect of drugs on cells cultured in
vitro in cell culture dishes. Cell cultures that are not exposed to the drug
(I) grow to a hypothetical 3-fold, or 300%, of their initial numbers on culturing in a growth-promoting media for
a fixed amount of time. Thus, these cells show 200 % growth, or 200% increase in viable cell count.
However, the cells exposed to the drug (II) have less number of viable cells on
culture under similar conditions for the same amount of time. The number of
viable cells in the drug-exposed culture dish depends on the drug
concentra-tion in a manner illustrated by curve II. The drug concentration, at
which the viable cell count after culture remains the same as the initial, that
is, at 100%, is defined as the total growth inhibitory (TGI) concentration. Drug
concentra-tion that halves the growth of cells in culture, that is, increase in
cell number to half of the levels seen without drug (which was 200%), is defined as the GI50
(growth inhibition to 50% level). Similarly, the concentration of drug that halves the viable
cell count from its initial level (which is 100%) is defined as LC50 (lethal concentration to 50% level). (With kind permission
from Springer Science+Business Media: Pharmaceutical Perspectives of Cancer Therapeutics,
Anticancer drug development, 2009, 49–92, Narang A.S. and Desai, D.S.)
in
the development pipeline through various metrics of their effectiveness. New
drug discovery research should meet multiple criteria, such as clini-cal
novelty, commercial opportunities, meeting unmet clinical need, and to build
intellectual property.
This
group also gets involved in identifying biomarkers and indicators of efficacy
and toxicity in preclinical species, for potential utilization as surrogates
for efficacy and toxicity assessment in the clinical studies. The discovery
biology group works closely with bioanalytical scientists, who get involved in
developing assays for compounds and physiological markers. The data generated
by these functions are critically analyzed by pharmaco-kineticists and
toxicologists to differentiate compounds for prioritization for advancement to
the next stages of the drug discovery and development process.
Chemistry
Working
closely with the discovery biology function, the discovery chemists provide an
array of purified compounds for investigation. Discovery chemists, typically
synthetic organic chemists who are involved in chemical synthesis,
purification, and characterization of new chemical entities (NCEs), form the
bulk of this function. This function is involved in identifying chemical
compounds that may have drug-like properties and would respond in desirable
ways (such as agonist or antagonist) on chosen enzymes, receptors, or other
targets. The drug design roles of these sci-entists frequently involve
molecular modeling of the target receptor and in silico assessments of receptor binding of potential chemical
structures and structural
modifications.
This
group gets closely involved in the identification of potential mea-sures of
efficacy, as well as in the pharmaceutical developability assess-ment of new
drug candidates to identify lead compounds that potentially maximize efficacy
and minimize potential developability risks and toxicity. Developing
structure–activity and structure–toxicity relationships is a key function of
this group of scientists. High-throughput synthesis and purifi-cation
technologies are commonplace in modern-day discovery chemistry laboratories.
The
discovery chemistry function works closely with analytical scientists to assess
the purity and identify of their compounds and with the discovery biology
function to assess the efficacy of animal, cell culture, or in vitro models. Working coherently,
discovery chemistry and discovery biology functions shortlist a few candidates
that enter preclinical assessment and optimization.
Pharmaceutics
Preclinical
pharmaceutical development involves optimization of potential drug candidates
for drug-like properties. Development of pharmaceuticals comprises diverse
group of scientists that assess both pharmaceutical dosage form developability
through a series of assessments of solubility as a func-tion of pH and in
various biorelevant fluids, chemical stability of the com-pound to various
stresses such as temperature and humidity, and polymorph stability. This
function also undertakes pharmacokinetic and toxicokinetic assessment, in close
collaboration with discovery biology colleagues, in one or more species, in an
effort to identify potential starting dose and effica-cious dose range for the
FIH administration and dose escalation clinical studies. Pharmaceutical
development scientists work closely with all func-tions involved in drug
characterization and administration to address three key aspects of any new
drug: stability, bioavailability, and manufacturability.
Preclinical
optimization, discovery chemistry, and discovery biol-ogy functions work
together as a team and may identify several molecules that go through
developability assessments and are compared against each other for an array of
desirable physicochemical and biological properties. The outcome of this
exercise is the identification of one lead candidate that provides optimum
balance of desirable attributes, while avoiding the unde-sirable ones. Often,
one or more backup candidates are identified in case any significant
undesirable observation (such as toxicity) is observed with the lead candidate.
Animal toxicology
As
drug candidates advance toward FIH, formal toxicological evaluation in animal
species is initiated. These studies are guided by the compound characteristics,
target therapeutic area and biological receptor, as well as regulations that
govern toxicity assessment in the animal species. Typically, toxicity is
studied in two species, one rodent and one nonrodent, with an intent to
identify target organs and organ systems that may exhibit toxicities at higher
doses. These studies involve increasing drug dosing and exposures in the animal
species until toxicity is observed and carefully documented. These studies
frequently combine plasma concentration assessment and biomarker studies, if a
biomarker has already been identified.
Toxicologists,
working with diverse teams of professionals, are involved in the design and
conduct of animal studies, as well as in the interpretation of observations.
The goal of toxicological assessments is to rule out any significant
toxicities, identify a starting dose for the clinical studies, and outline a
monitoring strategy for potential toxicities during clinical studies.
Toxicology studies seek to identify a maximum tolerated dose (MTD) that
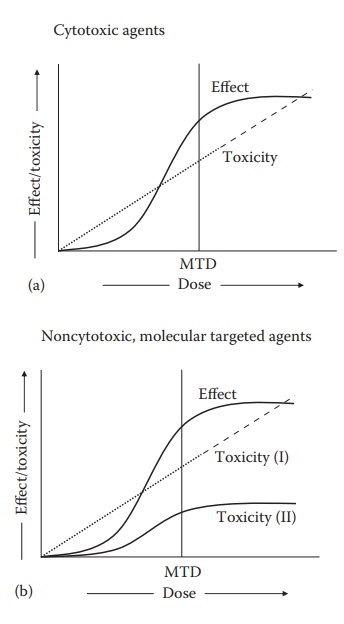
Figure 1.4 Hypothetical dose–effect and
dose–toxicity curves for cytotoxic (a) and noncytotoxic, molecularly targeted
anticancer (b) agents. The cytotoxic agents are known for their dose-dependent
toxicity, which closely follows the dose–effect curve. Noncytotoxic agents, on
the other hand, could have a linear dose–toxicity relationship similar to the
cytotoxic agents (I) or a nonlinear profile with dose–toxicity curve lower than
the dose–effect curve (II). MTD represents the maximum tolerated dose for the
cytotoxic agent. (With kind permission from Springer Science+Business Media: Pharmaceutical
Perspectives of Cancer Therapeutics, Anticancer drug development, 2009, 49–92,
Narang A.S. and Desai, D.S.)
presents
an acceptable balance of desired therapeutic effect and toxicity. Dose–effect
and dose–toxicity relationships are delineated to identify the balance of
effect and toxicity as a function of dose. As exemplified in Figure 1.4, these can be different for different
compounds, based on their mechanism of action and target specificity.
Clinical pharmacology
Although
no clinical studies are carried out with test candidates during drug discovery
stages, clinical pharmacologists get involved in understand-ing the emerging
profile of new drug candidates being screened. Aspects of drug discovery that
can impact later stages of drug development, such as the relevance of animal
models to human disease state and projected clini-cal doses or administration
profile (e.g., route or frequency of administra-tion), are critically assessed.
Analytical and bioanalytical sciences
Analytical
and bioanalytical sciences form the core indispensable compo-nent of all
functions involved in drug discovery and development. Analyses of drug content,
purity, and any changes during storage are an essential part of identification
of new chemical candidates through all stages of drug development, including
commercialization. All drugs are required by federal regulations to have
specification controls to ensure their identity, purity, and quality. The
analytical methodologies utilized for ensuring these attributes could be
spectroscopic or wet analytical techniques such as titrations and
chromatography. For example, an oral solid dosage form must be tested for drug
content, purity, water content, and drug release. A parenteral biologic drug
product must be tested for drug content, purity by different methods (charge or
size-based separation techniques), recep-tor binding, bioactivity, pH,
osmolality, and structure/isoforms. In addi-tion, all the starting (raw) materials,
intermediates for synthesis, and excipients used in formulations must be
rigorously tested to ensure these attributes and consistent quality across
different batches. The assurance of maintenance of all quality attributes of
the drug substance and drug product over the duration of storage (stability
testing) utilizes analytical testing at various time points under different
storage conditions (such as temperature, humidity, and light exposure). These
functions are carried out by analytical scientists.
Bioanalytical
sciences focus on analyses of drug content, metabolites, and any drug-related
substances (such as both the parent compound and prodrug in the case of prodrug
administration) in both animal and human studies. Bioanalytical sciences
present significant and unique challenges due to complex, multicomponent nature
of the biological fluids in which specific compounds must be tested—often
without rigorous separation— and the very low concentrations of the target
compounds (often in micro-molar quantities). These analyses are typically
carried out using highly sensitive analytical methods such as high-performance
or ultra-high per-formance liquid chromatography (HPLC/UPLC), followed by
tandem mass spectrometry (MS/MS).
All
analytical methods are required to be qualified and validated for a host of
criteria that ensure their robust and reproducible performance across
potentially multiple testing sites, laboratories, and personnel.
Regulatory sciences
All
drug products developed in modern biopharmaceutical settings are designed for
global patient populations. Government regulatory agencies that monitor and
control (regulate) the commercialization and utilization of drug products vary
by each country and as do their requirements for the testing and commercialization
of new drug products. As much harmoniza-tion of international regulations is
being advocated and implemented (e.g., by the International Council on
Harmonisation [ICH]), each country thus maintains its sovereign control over
access to its markets and the require-ments, which are often embedded in
historical idiosyncrasies and scientific elements that may be unique to each
region or subpopulation. The govern-ment regulatory agencies include, for
example, the federal Food and Drug Administration (FDA) in the United States
and the European Medicines Agency (EMA) for several countries in the European
Union; each of these countries do have their own drug regulatory agencies.
To
ensure access of new drug products to patient populations globally, regulatory
scientists work in the biopharmaceutical industry to proactively understand the
regulations of the targeted markets. The regulatory sciences have evolved into
a complex field that addresses not only the diversity of regulatory
requirements but also the variations in scientific perspectives and
understanding of different regions and countries. With ever-evolving analytical
methodologies, drug development paradigms, and accelerating scientific growth
in multiple disciplines involved in drug discovery and development, the
regulatory scientists also form the interface of biophar-maceutical companies
with the regulatory agencies and seek to educate and influence regulatory
policy to evolve with the times.
Related Topics
