Stage-gate process of new drug discovery and development
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Drug development
New drug discovery and development process can be divided into distinct sequential phases that evaluate and develop the drug-like characteristics of a potential new compound being considered a drug candidate.
Stage-gate process
of new drug discovery and development
New
drug discovery and development process can be divided into distinct sequential
phases that evaluate and develop the drug-like characteristics of a potential
new compound being considered a drug candidate (Figure
2.1). The progression of new drug candidates through various stages of
this sequential process depends on successful demonstration of drug-like
characteristics in each of these phases. Scientists working in a wide array of
disciplines are responsible for both characterization and enablement of
drug-like properties in new drug candidates throughout these stages of drug
development. These stages include early discovery, preclinical development,
FIH, registrational clinical studies, commercialization, and life cycle
management. There is a progressive reduction in the number of molecules that
progress through this stage-gate process, concurrent with increasing knowledge,
patient exposure, and understanding of the molecule (Figure
2.3).
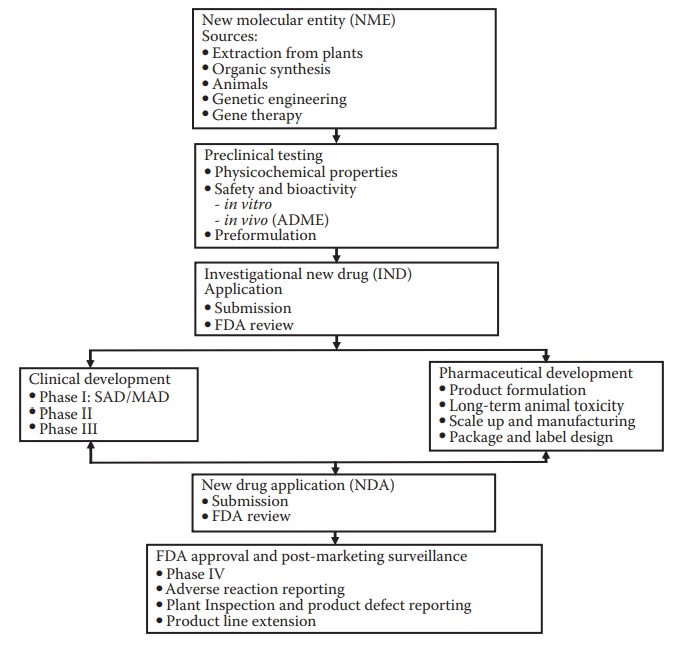
Figure 2.1 Stages of drug development. Drug discovery efforts lead to a new molecu-lar entity (NME) that is identified for development. The NME enters pre-clinical testing and development, which include efficacy and safety testing in animal species. This is followed by the submission of an investigational new drug (IND) application to the regulatory agency, such as the U.S. Food and Drug Administration (FDA). The drug then enters clinical trials: phase I, phase II, and phase III. Pharmaceutical development proceeds concurrent with clinical development and with the objectives of supporting the ongo-ing clinical studies (providing information, documentation, and the drug product for administration to the subjects) and preparation for commer-cialization of the product. Following successful clinical and pharmaceutical development, a data package is submitted to the FDA for approval; this is called the new drug application (NDA). Approval of the NDA is required for the commercialization of a new drug product. Several activities on the drug product continue after commercialization, such as adverse event monitoring, development of line extension products, and additional clinical trials to support label claims or expand target patient populations.
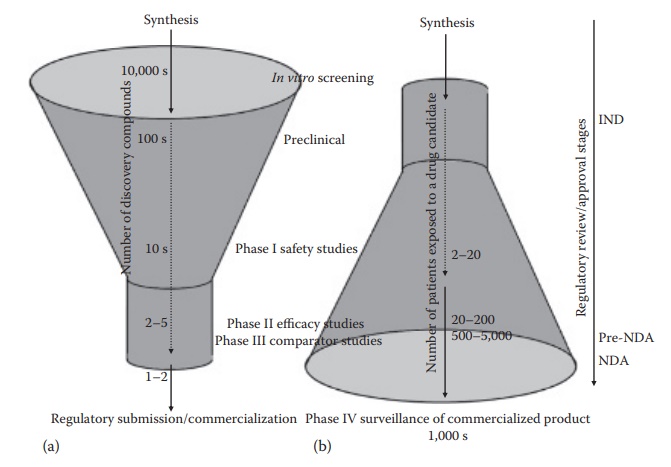
Figure 2.3 Number of compounds proceeding through various stage of drug development— an upright funnel (a), and the number of patients exposed to a given drug through its development—an inverse funnel (b). This figure also illustrates the steps where regulatory review and/or approval are required in the United States, such as the investigational new drug (IND) application submission before initiating phase I studies, a pre-NDA meeting with the FDA after the phase II studies, and new drug application (NDA) submission for drug approval for marketing after the completion of phase III studies. (With kind permission from Springer Science+Business Media: Pharmaceutical Perspectives of Cancer Therapeutics, Anticancer drug development, 2009, 49–92, Narang A.S. and Desai, D.S.)
These
stages are bounded by distinct boundaries, where the governance leadership of
an organization must make a decision whether to continue to invest in a
molecule or not. These are called decision-points
or stage-gate checkpoints. At these
points, interdisciplinary discussions that include both technical (e.g., developability risk) and nontechnical (e.g.,
commercial potential and competition) aspects identify risk–reward balance and
the path to commercialization of an asset, which feed into governance decision
for a given asset or a set of assets.
Preclinical development
Preclinical
development involves in-depth characterization of a few select potential drug
candidates, the NCEs or NMEs, before first human admin-istration. The
preclinical stage includes detailed physicochemical character-ization of the
compound and animal studies. This stage has the objectives of identifying
developability and clinical risks to the compound and of identifying a viable
development path to a commercial drug product. In addition, a critical decision
of the starting human dose is made based on the animal studies carried out
during the preclinical phase.
i. Physicochemical characterization
New
chemical entities are characterized for their pH–solubility profile,
pH–stability profile, solid form and form stability, excipient compatibility,
dissolution rate, photostability, supersaturation on pH transfer, and other
physicochemical characteristics. Prototype formulations are designed for use in
animal efficacy studies, toxicological assessment, and phase I clinical
studies. The ability to produce a commercially viable dosage form of the
compound is assessed at this stage.
ii. Efficacy studies
A
key goal of preclinical testing is to determine whether a compound exhib-its a
pharmacological activity and is reasonably safe for initial testing in humans.
Following identification of some lead compounds, the pharmaco-logical and
toxicological effects of these compounds are determined. These tests involve
the use of laboratory animals, cell culture, enzymes, and recep-tors, as well
as computer models. Animal testing may be carried out, for example, in
transgenic mice or other animals, that exhibit the pharmacology of a particular
disease state and/or drug target that defines the target patient population.
iii. Toxicology studies
Animal
toxicology assessment is carried out in at least two different animal species.
These toxicology studies are intended to assess the organs or organ systems in
which a particular compound tends to exhibit toxicity as well as to identify
the doses at which the toxicity appears. These animal studies are used to
understand how the drug is absorbed, distributed, and metabolized in the body;
ascertain its metabolites; and determine how quickly the drug is excreted from
the body. Pharmacokinetic and pharmacodynamic studies are conducted to analyze
the absorption, distribution, metabolism,
excre-tion, and toxicological effects
of the drug, commonly known as ADME/Tox prediction.
Animal
doses are translated into the FIH dose with normalization based on body weight
or surface area. Interspecies dose scaling for small-molecule compounds is
generally carried out using body weight as a metric, for example, mg dose per
kg body weight is kept constant across species. The metric for biologic
compounds, for example, therapeutic proteins and anti-bodies, is generally dose
per unit body surface area (e.g., mg dose per m2). The FIH doses are
typically a fraction of the lowest toxic dose observed in any animal species.
Clinical development
Clinical
development can be divided into four phases: phase I, II, III, and IV, with
progressively increasing number of subjects exposed to the drug. Such division,
through distinct clinical protocols, is intended to utilize the results of the
previous phase or subphase of the study to inform the design of the next phase
of the study.
Phase
I clinical studies are aimed at identifying dose-limiting toxicities,
toxicological profile, dose–exposure relationship, and drug pharmacokinet-ics
in a small group of healthy or patient volunteers. Phase II trials are designed
to confirm toxicological findings and generate information on pharmacologic
activity and pharmacokinetics. Phase II studies also aim to define the
appropriate dose for larger phase III trials. Phase III trials are the clinical
studies in a large group of patients. These studies are often also called
registrational clinical trials, since the results of these studies are
submitted (registered) to the FDA as a new drug application (NDA) or a
biologics license application (BLA) to gain regulatory approval for
commer-cialization of a new drug. Phase IV studies are postapproval clinical
studies that might include specific targeted studies that verify or expand the
cur-rent spectrum of claims on the label of the drug.
The
FIH clinical studies, also called phase I studies, are typically carried out in
a very small cohort of human subjects, often normal healthy volunteers. These
studies are intended to assess the safety of the compound. The drug is
administered at very low doses, based on the observations in animal studies.
The
doses are increased in single- or multiple-dose studies in a system-atic
fashion under close clinical observations to identify potential adverse events,
monitor drug pharmacokinetics and biochemical markers for those adverse events,
assess target engagement, and identify doses that may be utilized in subsequent
larger-scale phase II and phase III studies.
The
large late-stage clinical studies that form the basis of a drug’s approval are
considered registrational. Phase I
studies are typically car-ried out in healthy volunteers to assess toxicity and
define doses for human administration. Phase II studies may involve patients
and are uti-lized to identify adverse events in a broader population set and to
more closely define doses for the registrational studies. Registrational phase
III studies often involve comparison against a placebo or current
standard-of-care treatment. This may vary depending on the therapeutic category
and indication of the drug. For example, studies with cytotoxic antican-cer
drugs are not carried out in healthy volunteers, and placebo may not be used as
a comparator for patients with serious diseases. Increasingly, the demarcation
between different phases of clinical development are getting blurred with the
key defining criterion of clinical studies being restricted to
nonregistrational dose-escalation studies and registrational studies.
In
addition, several smaller, focused clinical studies are carried out at
dif-ferent points in time during drug development to support a drug product’s
labeling and interchangeability. For example, bioequivalence studies may be
carried out to bridge a phase I
formulation with a phase III formulation or to support a formulation change
during clinical development. Examples of other focused studies include food
effect evaluation, drug–drug interac-tion studies, and studies in special
populations such as in renally compro-mised patients.
The
larger-scale studies are carried out in an increasing number of sub-jects (Figure 2.3). As clinical trials progress, the dose
and dosing regimen are optimized to the effective levels that present an
acceptable toxicologic profile. In addition to the characterization of the
clinical profile of the drug candidate (such as the dose and frequency of
administration), these studies seek to differentiate the new drug candidate
from the existing therapeutic options for a given set of patients. Thus,
studies may be car-ried out to compare the efficacy and toxicity of the NCE
with the current standard of care.
Phase
IV studies involve postcommercialization monitoring of drug effects and are
designed to monitor a drug’s long-term safety and effective-ness, as well as to
uncover any rare but serious side effects that may not have been evident in
earlier, relatively smaller pool of healthy and patient volunteer studies. In
addition, studies may be carried out in special patient populations—such as
pediatrics, geriatrics, and renally compromised patients—to more closely define
the dosage and risk–benefit profile in those patient populations.
i. Phase I clinical trials
Phase
I studies are the FIH studies, which involve the first introduction of an IND
into humans. These studies are closely monitored and may be con-ducted in
patients (when ethically required, e.g., anticancer drugs), but are usually
conducted in healthy subjects. Phase I clinical trials are relatively short
(several months) and involve relatively less number of human volun-teers
(6–20). These studies are designed to identify a drug’s safety profile,
including the safe dosage range. The purpose of a phase I clinical trial is to
establish the tolerance of the drug in healthy human subjects at different
doses and define its pharmacologic effects. Thus, these studies often involve dose escalation within a clinical trial.
These
studies also involve measurement of plasma drug concentration to determine how
a drug is absorbed, distributed, metabolized, and excreted (ADME), as well as
the duration of its action. The purpose of a phase I clinical trial is to
establish a safe dosage range by determining the tolerance of the drug in
healthy human subjects at different doses and define its phar-macological
effects. Information about the pharmacokinetic profiles of the drug in humans
is used to design appropriate dosing regimens for the next phase of clinical
trials.
ii. Phase II clinical trials
Once
an experimental drug has proven to be safe and well tolerated in phase I
healthy volunteer studies, it is tested in patients in phase II studies. Phase
II clinical trials involve controlled studies on 100–300 volunteer patients to
assess the effectiveness of the drug for a particular indication(s) and
recon-firm toxicological profile. These studies are usually longer and larger
than phase I studies (1+ years). This phase of testing also helps determine the
common short-term side effects and risks associated with the drug.
Two
key aspects of late-stage clinical development are (1) randomization, and (2)
blinding.
·
Randomization: Most phase II
studies are randomized trials against a
control group; that is, one group of patients receives the experimen-tal drug,
while a second control group receives
the treatment that represents a current standard of care, or placebo. Placement
of the subject into the drug treatment or control group is done by random
assignment. The randomization of subject assignment is an important statistical
control to obviate any bias in study design.
·
Blinding: An important
methodology to avoid any bias during clinical testing in terms of patient perception and response is for the
patient to not know whether he or she is receiving the drug or the control
option. Such a clinical study in which the patient does not know the therapy
but the healthcare providers, including the physician, may know what is being
administered is called a single-blind study.
However,
often, the clinical studies are double-blinded;
that is, neither the patient nor the physician knows which patients are getting
the experi-mental drug. A double-blinding approach overcomes bias in
physician’s influence on patient perception and response as well as physician’s
report-ing of patient experience. The drug product manufacturer often carries
out double blinding by providing two look-alike medicinal products that are
only coded differently. In certain cases, the blinding may be carried out by
the on-site healthcare professional, such as the pharmacist, who prepares drug
products for administration and prepares the blinded labels.
Phase
II studies are designed to determine the correct dose and, thus, are often
referred to as dose-ranging studies. During phase II clinical trial, the final
dosage form is selected and developed for phase III trials. Phase II stud-ies
are sometimes divided into phase IIA and phase IIB studies. When defined
distinctly, phase IIA studies are considered exploratory evaluation of clinical
efficacy, with pharmacodynamics or biological activity as the primary
end-point. Phase IIB studies are then defined as the definite
dose-range-finding study that identifies doses for the large-scale,
registrational phase III studies.
iii. Phase III clinical trials
A
phase III clinical trial is an expanded controlled clinical trial of a drug’s
safety and efficacy in large and diverse patient populations. This phase
usually lasts several years and involves approximately 500–3,000 patients in
clinics and hospitals. Physicians monitor patients closely to determine
efficacy and identify adverse reactions. Phase III studies gather precise
information on the drug’s effectiveness for specific indications, determine
whether the drug produces a broad range of adverse effects than those exhibited
in the small study populations of phase I and II studies, and iden-tify the
best way of administrating and using the drug for the purpose intended. Phase
III studies also provide an adequate basis for extrapolating the results to the
general population and transmitting that information in the physician labeling.
iv. Evolving clinical development paradigms
In
cases where phase I studies involve patients and are adequately extensive and
detailed, phase II studies may be bypassed. Similarly, in cases where phase II
studies are extensive, they may be used as registrational studies, thus
bypassing the need for phase III studies. Such decisions are made on a
case-by-case basis, depending on the exact disease area and the therapeutic
profile of the investigational drug candidate.
As
mentioned earlier, the division of clinical studies into distinct phases is
based on the generation of distinct clinical protocols, with an intention to
utilize the results of the previous phase or subphase of the study to inform
the design of the next phase of the study. In cases where the decision tree for
how the previous phase of the study would influence the design of the
succeeding phase can be constructed before actually carrying out the first
phase of the study, clinical study designs for different phases can be made seamless. Such clinical studies continue
the treatment of a selected cohort of
patients enrolled in a phase II study into the extended phase III study, while
enrolling additional patients to meet the requirements of the number of
subjects needed for the phase III study.
In
addition, clinical study designs can be adaptive.
Adaptive clinical trial designs allow changes in the design or analyses while
the study is in prog-ress. These changes are guided by the accumulated data at
an interim point in the trial. Such designs make the clinical study more
efficient in their abil-ity to reduce the duration or the number of subjects
required for the study.
Related Topics
