Influential factors in dosage form design
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Pharmaceutical considerations
Each drug substance has intrinsic chemical and physical characteristics that must be considered before the development of its pharmaceutical formulation.
Influential
factors in dosage form design
Each
drug substance has intrinsic chemical and physical characteristics that must be
considered before the development of its pharmaceutical formulation. Among these
characteristics are the particle size, surface area, the drug’s solubility, pH,
partition coefficient, dissolution rate, physical form, and stability. All
these factors are discussed below, except the particle size and dissolution
rate, which will be discussed in the next chapter.
Molecular size and volume
Molecular
size and volume have important implications for drug absorp-tion. Tight
junctions can block the passage of even relatively small molecules, whereas gap
junctions are looser. Molecules up to 1,200 Da can pass freely between cells;
however, larger molecules cannot pass through gap junctions. Drug diffusion in
simple liquid is expressed by Stokes–Einstein equation:
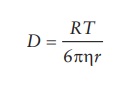
D
= RT/6πηr
where:
D is the diffusion of
drugs
R is the gas constant = 8.313 JK−1mol−1
T is the temperature
(Kelvin)
η is the solvent
viscosity
r is the solvated
radius of diffusing solute
Since
volume (V) = (4/3) πr3, the above
equation suggests that drug diffu-sivity is inversely proportional to the
molecular volume. Molecular volume is dependent on molecular weight,
conformation, and heteroatom content. Molecules with a compact conformation
will have a lower molecular vol-ume and thus a higher diffusivity. As shown in Figure 3.1, the diffusion and permeability of the
endothelial monolayer to molecules decreased with increasing molecular weight.
A
drug must diffuse through a variety of biological membranes after
administration into the body. In addition, drugs in many controlled-release
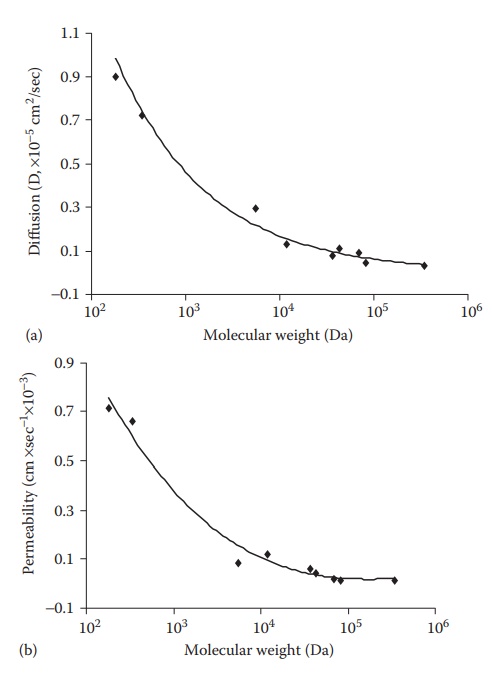
Figure 3.1 Diffusion (a) and permeability
(b) of compounds with different molecular weight across an endothelial
monolayer at 37°C.
systems
must diffuse through a rate-controlling membrane or matrix. The ability of a
drug to diffuse through membranes is a function of its molecu-lar size and
volume. For drugs with a molecular weight greater than 500, diffusion in many
polymeric matrices is very small. Lipinski devised the so-called Rule of 5, which refers to drug-like
properties of molecules. It states that poor oral absorption is more likely
when the drug molecule has:
·
More than five hydrogen-bond donors (–OH groups or –NH
groups).
·
A molecular weight greater than 800.
·
A log P > 5.
·
More than 10H-bond acceptors.
However,
this rule is not applicable to the compounds that are substrates for
transporters.
Drug solubility and pH
Pharmacological
activity is dependent on solubilization of a drug sub-stance in physiological
fluids. Therefore, a drug substance must pos-sess some aqueous solubility for
systemic absorption and therapeutic response. Enhanced aqueous solubility may
be achieved by forming salts or esters, by chemical complexation, by reducing
the drug’s particle size (i.e., micronization), or by creating an amorphous
solid. One of the most important factors in the formulation process is pH, as
it affects solubility and stability of weakly acidic or basic compounds.
Changes in pH can lead to ionization or salt formation. Adjustment in pH is
often used to increase the solubility of ionizable drugs, because the ionized
molecular species have higher water solubility than their neutral species.
According to Equations 3.1 and 3.2, the total solubility, ST, is the function of intrin-sic solubility, S0, and the difference
between the molecule’s pK a
and the solution pH. The intrinsic solubility is the solubility of the neutral
spe-cies. Weak acids can be solubilized at pHs below their acidic pKa, whereas weak bases can be
solubilized at pHs above their basic pKa.
For every pH unit away from the pKa,
the weak acid–base solubility increases 10-fold. Thus, solubility can be
achieved as long as the formulation pH is at least 3 units away from the pKa. Adjusting solution pH is
the simplest and most common method to increase water solubility in injectable
products.
For
a weak acid ST = S0(1
+ 10pH−p Ka ) (3.1)
For
a weak base ST = S0(1
+ 10p Ka −pH) (3.2)
Unlike
a weak acid or base, the solubility of a strong acid or base is less affected
by pH. The drugs without ionizable groups are often solubilized by the
combination of an aqueous solution and water-soluble organic solvent/
surfactant. Frequently, a solute is more soluble in a mixture of solvents than
in one solvent alone. This phenomenon is known as cosolvency, and the solvents that in combination increase the
solubility of the solute are called cosolvents.
The addition of a cosolvent can increase the solubility of hydro-phobic
molecules by reducing the dielectric
constant, which is a measure of the influence by a medium on the energy
needed to separate two oppositely charged bodies. Some of the cosolvents
commonly being used in pharmaceu-tical formulations include ethyl alcohol,
glycerin, sorbitol, propylene glycol, and polyethylene glycols (PEGs).
Polyethylene glycol 300 or 400, propyl-ene glycol, glycerin, dimethylacetamide
(DMA), N-methyl 2-pyrrolidone (NMP),
dimethyl sulfoxide (DMSO), Cremophor, and polysorbate 80 are often used for
solubilization of drugs that have no ionizable groups. As shown in Figure 3.2, the solubility of phenobarbital is, for
example, signifi-cantly increased in a mixture of water, alcohol, and glycerin
compared with
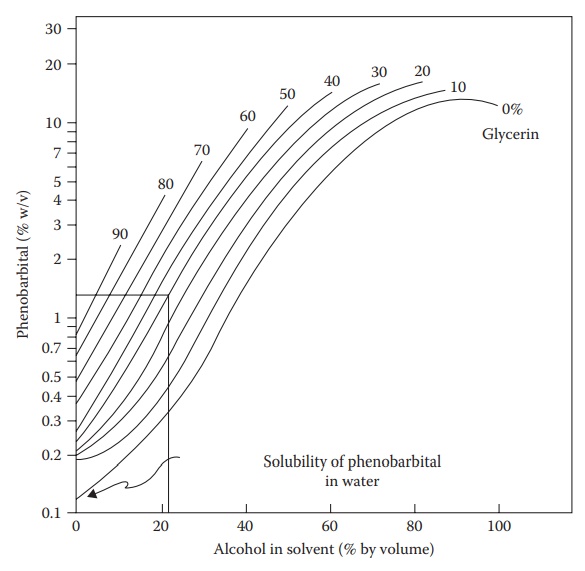
Figure 3.2 Effect of cosolvents on the
solubility of phenobarbital in a mixture of water, alcohol, and glycerin at 25°C. (Reproduced from Krause, G.M.
and Gross, J.M., J. Am. Pharm. Assoc. Ed., 40, 137, 1951. With permission.)
However, the use of cosolvents often leads to the
precipitation of the drug on dilution during the administration of the drug
solution into the body, resulting in pain or tissue damage.
Excipients
that solubilize a molecule via specific interactions, such as complexation with
a drug molecule in a noncovalent manner, lower the chemical potential of the
molecules in solution. These noncovalent solubility-enhancing interactions are the
basis of the phenomenon that like
dissolves like and include van
der Waals forces, hydrogen bonding, dipole–dipole, ion–dipole interactions, and, in certain cases, favorable
electromagnetic interactions. Solutes dissolve better in solvents of similar polarity.
Therefore, to dissolve a highly polar or ionic compound, one should use a
solvent that is highly polar or has a high dielectric constant. On the
contrary, to dissolve a drug that is nonpolar, one should use a solvent that is
relatively nonpolar or has a low dielectric constant.
Table 3.2 Water solubility of different
substituent
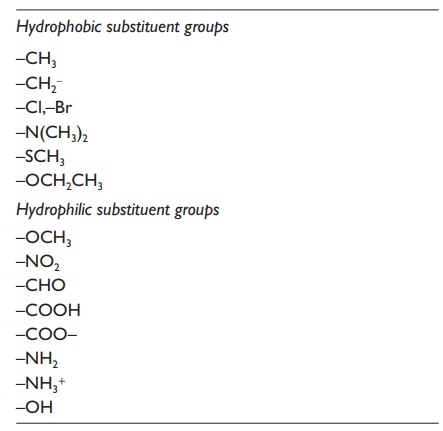
Drug
solubility can also be enhanced by altering its structure; this is one basis
for the use of prodrugs. A prodrug is
a drug that is therapeutically inactive when administered but becomes activated
in the body by chemical or enzymatic processing. The addition of polar groups,
such as carboxylic acids, ketones, and amines, can increase aqueous solubility
by increasing the hydrogen bonding and the dipole–dipole interaction between
the drug molecule and the water molecules. Table 3.2
lists different substituents that will have significant influence on the water
solubility of drugs. Substituents can be classified as either hydrophobic or
hydrophilic, depending on their polarity. The position of the substituents on
the molecule can also influence its effect.
Lipophilicity and partition coefficient
Partitioning
is the ability of a compound to distribute in two immiscible liquids. When a
weak acid or base drug is added to two immiscible liq-uids, some drug goes to
the nonpolar phase and some drug goes to the aqueous layer. Because like dissolves like, the nonpolar
species migrates (partitions) to the nonpolar layer and the polar species
migrate to the polar aqueous layer.
To
produce a pharmacologic response, a drug molecule must first cross a bio-logic
membrane, which acts as a lipophilic barrier to many drugs. Since passive
diffusion is the predominant mechanism by which many drugs are transported, the
lipophilic nature of the molecules is important. A drug’s partition
coeffi-cient is a measure of its distribution in a lipophilic–hydrophilic phase
system, and it indicates the drug’s ability to penetrate biologic multiphase
systems. The octanol–water partition coefficient is commonly used in
formulation develop-ment and is defined as:
P = Concentration of drug in octanol or
nonpolar phase / Concentration of drug in water or polar phase
For
an ionizable drug, the following equation is applicable:
P
= Concentration of drug in octanol or nonpolar phase / (1 − α)(Concentration of
drug in water of polar phase)
In
this equation, α is equal to the
degree of ionization. The concentration in aqueous phase is estimated by an
analytical assay, and concentration in octanol or other organic phases is
deduced by subtracting the aque-ous amount from the total amount placed in the
solvents. Partition coef-ficient can be used for drug extraction from plants or
biologic fluids, drug absorption from dosage forms, and recovery of antibiotics
from fermenta-tion broth.
The
logarithm of partition coefficient (P)
is known as log P. Log P is a measure of lipophilicity and is
used widely, since many pharmaceutical and biological events depend on
lipophilic characteristics. Often, the log P
of a compound is quoted. Table 3.3 lists the
log P values of some representative
compounds. For a given drug:
If
log P = 0, there is equal drug distribution
in both phases.
If
log P > 0, the drug is lipid
soluble.
If
log P < 0, the drug is water
soluble.
In
general, the higher the log P, the
higher the affinity for lipid membranes and thus the more rapidly the drug
passes through the membrane via pas-sive diffusion. However, there is a
parabolic relationship between log P
and drug activity when percentage of drug absorption is plotted against log P values (Figure
3.3). The parabolic nature of bioactivity and log P values is due to the fact that drugs with high log P values, protein bind-ing, low
solubility, and binding to extraneous sites cause them to have a
Table 3.3 Log P values of representative drugs
Drug : Log P
Acetylsalicylic acid 1.19
Amiodarone 6.7
Benzocaine 1.89
Bromocriptine 6.6
Bupivacaine 3.4
Caffeine 0.01
Chlorpromazine 5.3
Cortisone 1.47
Desipramine 4.0
Glutethimide 1.9
Haloperidol 1.53
Hydrocortisone 4.3
Indomethacin 3.1
Lidocaine 2.26
Methadone 3.9
Misoprostol 2.9
Ondansetron 3.2
Pergolide 3.8
Phenytoin 2.5
Physostigmine 2.2
Prednisone 1.46
Sulfadimethoxine 1.56
Sulfadiazine 0.12
Sulfathiazole 0.35
Tetracaine 3.56
Thiopentone 2.8
Xamoterol 0.5
Zimeldine 2.7
lower
bioactivity. Decrease in activity is due to the limitation in solubility beyond
a certain log P value. If a drug is
too lipophilic, it will remain in the lipid membrane and not partition out
again into the underlying aque-ous environment. On the other hand, very polar
compounds with very high log P values
are not sufficiently lipophilic to be able to pass through lipid membrane
barriers.
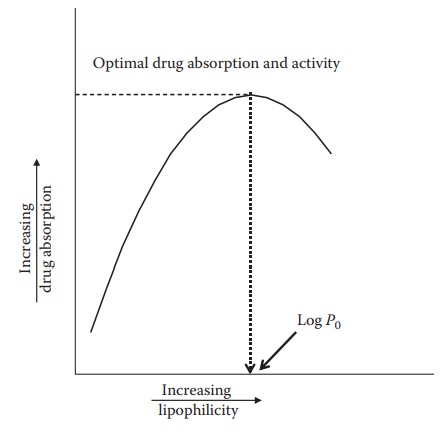
Figure 3.3 Relationship between drug absorption and log P. Decrease in the drug
absorp-tion beyond a certain log P value is probably due to its binding to
plasma proteins, reduction in free drug levels, or its binding to extraneous
sites.
Polymorphism
The
capacity of a substance to exist in more than one solid state forms is known as
polymorphism, and the different
crystalline forms are called polymorphs.
If the change from one polymorph to another is reversible, the process is enantiotropic.
However, if the transition from a metasta-ble to a stable polymorph is
unidirectional, the system is monotropic.
Polymorphic forms may exhibit detectable differences in some or all of the
following properties: melting point, dissolution rate, solubility, and
stabil-ity. Drug substances can be amorphous (i.e., without regular molecular
lattice arrangements), crystalline (which are more oriented or aligned),
polymorphic, anhydrous, or solvated. An important form in the formula-tion is
the crystal or amorphous form of the drug substance. Many drug substances can
exist in more than one crystalline form, with different lat-tice arrangements.
This property is termed polymorphism. Drugs may undergo a change from one
metastable polymorphic form to a more stable polymorphic form. Various drugs
are known to exist in different polymor-phic forms (e.g., cortisone and
prednisolone). Polymorphic forms usually exhibit different physicochemical
properties, including melting point and solubility, which can affect the
dissolution rate and thus the extent of their absorption. The amorphous form of
a compound is always more soluble than the corresponding crystal form. Changes
in crystal characteristics can influence bioavailability and stability and thus
can have important impli-cations for dosage form design. For example, insulin
exhibits a differing degree of activity, depending on its state. The amorphous
form of insulin is rapidly absorbed and has short duration of action, whereas
the large crys-talline product is more slowly absorbed and has a longer
duration of action.
Stability
The
chemical and physical stability of a drug substance alone and when combined
with formulation components is critical to preparing a successful
pharmaceutical product. Drugs containing one of the following functional groups
are liable to undergo hydrolytic degradation: ester, amide, lactose, lactam,
imide, or carbamate. Drugs that contain ester linkages include acetylsalicylic
acid, physostigmine, methyldopa, tetracaine, and procaine. For example, the
hydrolysis of acetylsalicylic acid (commercially known as aspirin) is
represented in Figure 3.4. Aspirin is
hydrolyzed to salicylic acid and acetic acid.
Nitrazepam,
chlordiazepoxide, penicillins, and cephalosporins are also susceptible to
hydrolysis. Several methods are available to stabilize drug solutions that are
susceptible to hydrolysis. For example, protection against moisture in
formulation, processing, and packaging may prevent decompo-sition. Suspending
drugs in nonaqueous solvents such as alcohol, glycerin, or propylene glycol may
also reduce hydrolysis.
After
hydrolysis, oxidation is the next
most common pathway for drug degradation. Drugs that undergo oxidative
degradation include morphine, dopamine, adrenaline, steroids, antibiotics, and
vitamins. Oxidation can be minimized by storage under anaerobic conditions.
Since it is very dif-ficult to remove all of the oxygen from a container,
antioxidants are often added to formulations to prevent oxidation.
Excipients used to prepare a
solid dosage can also affect the drug’s stability,
possibly by increasing the moisture content of the preparation. Excipients,
such as starch and povidone, have very high water contents.
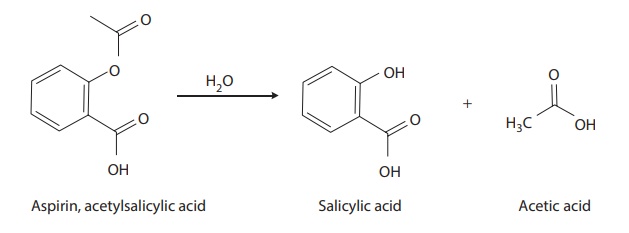
Figure 3.4 Hydrolysis of aspirin.
Acetylsalicylic acid (aspirin) is hydrolyzed to salicylic acid and acetic acid.
Povidone
contains about 28% equilibrium moisture at 75% relative humid-ity. However, the
effect of this high moisture content on the stability of a drug will depend on
how strongly it is bound and whether the moisture can come in contact with the
drug. Effects of tablet excipients on drug decom-positions are widely reported
in the literature. For an example, magnesium trisilicate is known to cause
increased hydrolysis of aspirin in the tablet because of its high moisture
content.
pKa/Dissociation constants
Many
drug substances are either weak acids or weak bases and thus undergo a
phenomenon known as dissociation when dissolved in liquid medium. If this
dissociation involves a separation of charges, then there is a change in the
electrical charge distribution on the species and a separation into two or more
charged particles, or ionization. The extent of ionization of a drug has an
important effect on the formulation and pharmacokinetic profiles of the drug.
The extent of dissociation or ionization is dependent on the pH of the medium
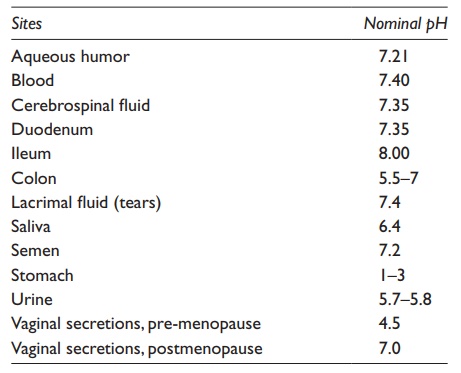
containing the drug. Table 3.4 lists the normal
pHs of some organs and body fluids, which are used in the prediction of the
percentage ionization of drugs in vivo.
In a formulation, often, the vehicle is adjusted to a certain pH to obtain a
certain level of ionization of the drug for solubility and stability. The
extent of ionization of a drug has a strong effect on its extent of absorption,
distribution, and elimination.
Acids
tend to donate protons to a system at pH greater than 7, and bases tend to
accept protons when added to acidic system (i.e., at pH < 7). Many drugs are
weak acids or bases and therefore exist in both unionized and
Table 3.4 Nominal pH values of some body
fluids and sites
The fraction of a drug that
is ionized in solution is given by the dissociation constant (Ka) of the drug. Such
dissociation constants are conveniently expressed in terms of pKa values for both acidic and
basic drugs. For a weak acidic drug, HA (e.g., aspirin and phenylbutazone), the
equilibrium is presented by:
HA
↔ H++A−
The
symbol (↔) indicates that
equilibrium exists between the free acid and its conjugate base. According to
the Bronsted–Lowry theory of acids
and bases, an acid is a substance that will donate a proton and a base is a
sub-stance that will accept a proton. Based on this theory, the conjugate base
A− may accept a proton and revert to the free acid. Therefore, the
dissocia-tion constant for this reaction is:
K1[HA] ↔ K2[H+][A−]
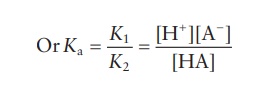
Taking
logarithms of both sides:
log
Ka = log[H+ ] + log[A− ] − log[HA]
The
signs in this equation may be reversed to give the following equation:
-log
Ka = − log[H+ ] − log[A− ] + log [HA]
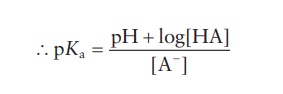
This
is a general equation applicable for any weakly acidic drugs.
Similarly,
for a weak basic drug (e.g., chlorpromazine) →B + H+ = BH+
∴ pKa = pH + log[BH + ] / [ B] for a
weakly basic drug.
These
equations are known as Henderson–Hasselbalch
equations. This equation describes the derivation of pH as a measure of
acidity (using pKa) in
biologic and chemical systems. The equation is also useful for estimat-ing the
pH of a buffer solution and finding the equilibrium pH in acid– base reactions.
Bracketed quantities such as [Base]
and [Acid] denote the molar
concentration of the quantity enclosed. Based on these equations, it is
apparent that the pKa is
equal to the pH when the concentration of the ionized and nonionized species is
equal (i.e., log1 = 0). It is therefore
impor-tant to realize that a compound is only 50% ionized when the pKa is equal to the pH.
Ionization constants are usually expressed in terms of pKa values for both acidic and basic drugs. The strength
of acid is inversely related to the magnitude of its pKa. The lower is the pKa, the stronger is the acid. Conversely, the strength
of a base is directly related to the magnitude of its pKa. The pK a
of a strong base is high. The pKa
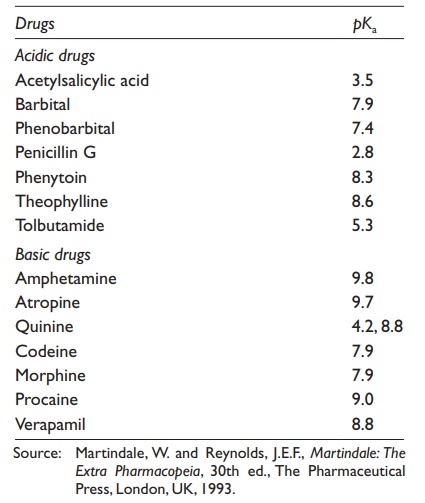
values of a series of drugs are listed in Table 3.5.
Acidic drugs are completely unionized at pHs up to 2 units below their pKa and are completely ionized
at pHs greater than 2 units above their pKa.
Conversely, basic drugs are completely ionized at pH up to 2 units below their
pKa and are completely
unionized when the pH is greater than 2 units above their pKa. Both types of drugs are exactly 50% ionized at their
pK a values. Some drugs
can donate or accept more than one proton, and so, they may have several pKa values.
For
either weak acid or base, the ionized species, BH+ and A−,
have very low solubility and are virtually unable to permeate membrane, except
where specific transport mechanisms exist. The lipid solubility of the
uncharged drugs will depend on the physicochemical properties of the drug.
Proteins
and peptides contain both acidic (−COOH) and basic (−NH2) groups.
The pKa values of
ionizable groups in proteins and peptides can be significantly different from
those of the corresponding groups when they
Table 3.5 pKa values of typical acidic and basic drugs
Therefore, these compounds are often referred to as
amphoteric in nature. The pH of a solution determines the net charge on the
molecule and ultimately the solubility. Since water is a polar solvent and
ionic species are more water soluble than the nonionic ones, a conjugate acid
(BH+) and a conjugate base (A−) are generally more water soluble
than the corresponding free base (B) or free acid (HA).
Example 3.1
The
pKa value of aspirin,
which is a weak acid, is about 3.5. What are the ratios of unionized and
ionized forms of this drug in the stomach (pH 2) and in the plasma (pH 7.4)?
Why does aspirin often cause gas-tric bleeding?
Answer:
According
to the Henderson–Hasselbalch equation,
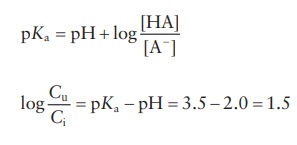
where
Cu is the concentration of
unionized drug and Ci is
the concen-tration of ionized drug.
∴ Cu/ Ci = antilog1.5 = 31.62:1
In
the plasma,
log
Cu/ Ci = pKa − pH = 3.5 − 7 .4 = −3 .9
∴ Cu/Ci = antilog( − 3.9) = 1.259 ×10−4 :
1
Therefore,
most of the administered aspirin remains unionized in the stomach, and thus, it
is rapidly taken up by the stomach, leading to gastric bleeding.
Degree of ionization and pH-partition theory
For
a drug to cross a membrane barrier, it must normally be lipid solu-ble to get
into the biological membranes. The ionized forms of acidic and basic drugs have
low lipid:water partition coefficients compared with the coefficients of the
corresponding unionized molecules. Lipid membranes are preferentially permeable
to the latter species. Thus, an increase in the fraction of a drug that is
unionized will increase the rate of drug trans-port across the lipid membrane.
This phenomenon can be explained by the pH-partition
theory, which states that drugs are absorbed from biological membranes by passive diffusion,
depending on the fraction of the union-ized form of the drug at the pH of that
biological membrane. Based on the Henderson–Hasselbalch
equation, the degree of ionization of a drug will depend on both its pKa
value and the solution’s pH.
The
gastrointestinal (GI) tract acts as a lipophilic barrier, and thus, ionized
drugs, which will be more hydrophilic, will have minimal membrane trans-port
compared with the unionized form of the drug. The solution pH will affect the
overall partition coefficient of an ionizable substance. The pI of the molecule is the pH at which
there is a 50:50 mixture of conjugate acid–base forms. The conjugate acid form
will predominate at a pH lower than the pKa,
and the conjugate base form will be present at a pH higher than the pKa.
Limitations of pH-partition theory
Although
the pH-partition theory is useful, it often does not hold true. For example,
most weak acids are well absorbed from the small intes-tine, which is contrary
to the prediction of the pH-partition hypothesis. Similarly, quaternary
ammonium compounds are ionized at all pHs but are readily absorbed from the GI
tract. These discrepancies arise because pH-partition
theory does not take into account the following:
·
The small intestine has a large epithelial surface area for
drug absorp-tion to take place. This large epithelial area results from mucosa,
villi, and microvilli (Figure 3.5). The large
mucosal surface area compen-sates for ionization effects.
·
Drugs have a relatively long residence time in the small
intestine, which also compensates for ionization effects.
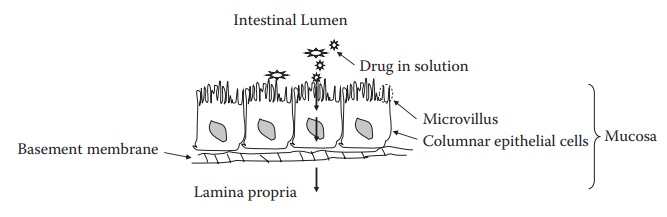
Figure 3.5 Drug absorption across small
intestine. The small intestine has a large epi-thelial surface area due to
mucosa, villi, and microvilli. This large surface area compensates the effect
of drug ionization on its absorption across the small intestine and invalidates
pH-partition theory of drug absorption.
·
Charged drugs, such as quaternary ammonium compounds and
tetra-cyclines, may interact with oppositely charged organic ions, resulting in
a neutral species, which is absorbable.
·
Some drugs are absorbed via active pathways.
· Many more.
Related Topics