Review questions and answers
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Pharmaceutical considerations
Pharmaceutical Drugs and Dosage: Pharmaceutical considerations - Review questions and answers
Review questions
3.1 Which of
the following statements is FALSE?
A. The partition
coefficient is the ratio of drug solubility in n-octanol to that in water.
B. Absorption of a weak
electrolyte drug does not depend on the extent to which the drug exists in its
unionized form at the absorp-tion site.
C. Amorphous forms of
drug have faster dissolution rates than crys-talline forms.
D. All of the above.
3.2 The pH
of a buffer system can be calculated with:
A. Henderson–Hasselbalch
equation
B. Noyes–Whitney
equation
C. Michaelis–Menten
equation
D. Yang’s equation
E. All of the above
3.3 Indicate which of
the following statement are TRUE and which are FALSE:
A. Henderson–Hasselbalch
equation describes the effect of physical parameters on the stability of
pharmaceutical suspensions.
B. The passive
diffusion rate of hydrophobic drugs across biological membranes is higher than
that of hydrophilic compounds.
C. Factors influencing
dosage form design do not include drug solubility and pH but include partition
coefficient and pKa
values.
D. Drug solubility can
be enhanced by salt formation, use of cosolvent, complex formation, and
micronization.
3.4 A. What is the
difference between drug adsorption and drug absorption?
B. Describe the pH-partition theory and its limitation in
relation to drug absorption across the GI tract.
C. Compare any two compounds differing in the following
character-istics and suggest which one would be absorbed more efficiently and
why:
i. A water-insoluble compound versus a highly soluble
compound.
ii.
A low molecular weight compound versus a high molecular weight compound.
3.5 A. Why do we need to
formulate a drug into a pharmaceutical dosage form?
B. Define partition coefficient and log P.
C. Define electrolytes and nonelectrolytes.
3.6 A. Enlist eight
intrinsic characteristics of a drug substance that must be considered before
the development of its pharmaceutical formulation.
B. Enlist two limitations of pH-partition theory.
3.7 Define
pH-partition theory. The pKa value of aspirin, which
is a weak acid, is
about 3.5. What are the ratios of ionized and unionized forms of the drug in
the stomach (pH 2) and in the plasma (pH 7.4)? Why does aspirin often cause
gastric bleeding?
3.8 Enlist
six physicochemical properties of a drug that influence absorp-tion. How can
the physicochemical properties be improved to increase drug absorption?
3.9 The pKa of pilocarpine is 7.15 at 25°C. Compute the mole percentage of free base present at
25°C and a pH of 7.4.
3.10 Calculate
the percentage of cocaine existing as the free base in a solu-tion of cocaine
hydrochloride at pH 4.5 and pH 8.0. The pKa
of cocaine is 5.6.
3.11 For a
weak acid with a pKa of 6.0, calculate the
ratio of acid to salt at pH 5.
Answers:
3.1 B. According to the pH-partition
theory, absorption of a weak elec-trolyte drug depends on the extent to which
the drug exists in its unionized form at the absorption site. However, the
pH-partition theory often does not hold true, as most weakly acidic drugs are
well absorbed from the small intestine because of the large epithe-lial surface
areas of the organ.
3.2 A. The
Henderson–Hasselbalch equation for a weak acid and its salt is represented as pH = pKa + log [salt]/[acid], where pKa is the negative log of the
dissociation constant of a weak acid and [salt]/ [acid] is the ratio of the
molar concentration of salt and acid used to prepare a buffer.
3.3 A. False
B. True
C. False
D. True
3.4 A. Adsorption is different from absorption, which implies pen-etration
through organs and tissues. The degree of adsorption depends on the chemical
nature of the adsorbent and the adsor-bate, surface area of the adsorbent,
temperature, and partial pres-sure of the adsorbed gas. Adsorption can be
physical or chemical in nature.
B.
The pH-partition theory states that
drugs are absorbed from the biological membranes by passive diffusion,
depending on the fraction of unionized form of the drug at the pH of that
biologi-cal membrane. Their degree of ionization depends on both their pKa and the solution pH. The
GI tract acts as a lipophilic bar-rier and thus ionized drugs will have minimal
membrane perme-ability compared to unionized form of the drug. The solution pH
will affect the overall partition coefficient of an ionizable substance. The pKa of the molecule is the pH
at which there is a 50:50 mixture of conjugate acid–base forms. The conjugate
acid form will predominate at a pH lower than the pKa, and the conjugate base form will be present at a pH
higher than the pKa. The
following Henderson–Hasselbalch equations
describe the relationship between ionized and nonionized species of a weak
electrolyte:
Weakly
Acidic Drugs
pH
= p Ka + log[A− ]/[HA]
Weakly
Basic Drugs
pH
= pKa + log[ B− ]/[BH+]
The
pH-partition theory often does not hold
true. For example, most weak acids are well absorbed from the small
intestine, which is contrary to the prediction of the pH-partition hypothesis.
Similarly, quaternary ammonium compounds are ionized at all pHs but are readily
absorbed from the GI tract. These discrep-ancies arise because the pH-partition theory does not take into account the following:
·
Large epithelial surface areas of the small intestine compensate
for ionization effects.
·
Long residence time in the small intestine also compensates
for the ionization effects.
·
Charged drugs, such as quaternary ammonium compounds, may
interact with oppositely charged organic ions, resulting in a neutral species,
which is absorbable.
·
Some drugs are absorbed via active pathways.
C.
i. A highly water-soluble compound will be absorbed poorly as compared to
lipophilic compounds.
ii. A low-molecular weight compound will be absorbed better
than a high-molecular weight compound.
3.5 A.
Besides providing the mechanism for the safe and convenient deliv-ery of
accurate dosage, we need to formulate a drug into pharma-ceutical dosage forms
for the following additional reasons: (i) to protect the drug substance from
the destructive influence of atmo-spheric oxygen or humidity;(ii) to protect
the drug substance from the destructive influence of gastric acid after oral
administration; (iii)
to conceal the bitter, salty, or offensive taste or odor of a drug substance;
(iv) to provide liquid preparations of substances that are either insoluble or
unstable in the desired vehicle; (v) to pro-vide rate-controlled drug action;
and (vi) to provide site-specific drug delivery.
B.
A drug’s partition coefficient is a measure of its distribution in a
lipophilic–hydrophilic phase system and indicates its ability to penetrate
biological membranes. The octanol–water parti-tion coefficient is used in the
formulation development and is defined as P
= (concentration of drug in octanol or nonpolar solvent)/(concentration of drug
in water polar solvent). The log-arithm of partition coefficient (P) is known as log P. The value of log P is
a measure of lipophilicity and is used widely because many pharmaceutical and
biological events depend on lipophilic characteristics.
C.
Nonelectrolytes are substances that do not form ions when dissolved in water.
Their aqueous solutions do not conduct an electric current. Electrolytes are
substances that form ions in solution. As a result, their aqueous solutions
conduct electric current. Electrolytes are characterized as strong or weak.
Strong electrolytes (e.g., sodium chloride and hydrochloric acid) are
completely ionized in water at all concentrations. Weak electrolytes (e.g.,
aspirin and atropine) are partially ionized in water.
3.6 A. Eight intrinsic
characteristics of a drug substance that must be considered before the
development of its pharmaceutical formula-tion are the following:
Drug solubility and
pH:
A drug substance must possess some aqueous
solubility for systemic absorption and therapeu-tic response. Enhanced aqueous
solubility may be achieved by forming salts or esters, by chemical
complexation, or by reducing the drug’s particle size. The pH affects
solubility and stability. Cosolvents, complexation, micronization, and solid
dispersion are used to improve aqueous solubility.
Partition
coefficient:
The partition coefficient of a drug is
a measure of its distribution in a lipophilic–hydrophilic phase system and
indicates its ability to penetrate biological membranes.
Dissolution rate: The speed at which
a drug substance dis-solves in a medium is called its dissolution rate.
Polymorphism: Polymorphic forms
exhibit different physico-chemical properties, including melting point and
solubility, which can affect the dissolution rate and thus the extent of its
absorption.
Stability: The chemical and
physical stability of a drug sub-stance alone, and when combined with
formulation compo-nents, is critical to prepare a successful pharmaceutical
product. For drugs susceptible to oxidative decomposition, the addition of
antioxidant stabilizing agents to the formulation may be required to protect
potency. For drugs destroyed by hydroly-sis, protection against moisture in
formulation, processing, and packaging may be required to prevent
decomposition.
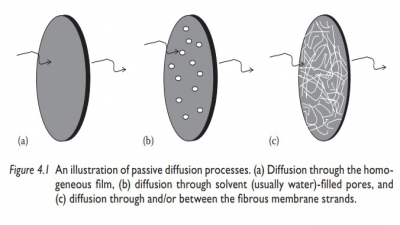
Membrane
permeability:
To produce a biological response, the
drug molecule must first cross a biological membrane. The biological membrane
acts as a lipid barrier to most drugs and permits the absorption of
lipid-soluble substances by passive diffusion, whereas lipid-insoluble drugs
can diffuse across the barrier only with considerable difficulty.
Partition
coefficient:
The octanol–water partition coefficient is
used in formulation development. P
= (concentration of drug in octanol)/(concentration of the drug in water).
pKa/dissociation constants: The extent of
ionization or dissocia-tion is dependent on the pH or the medium containing the
drug.
B. The pH-partition
theory often does not hold true, as most weakly acidic drugs are well absorbed
from the small intestine, possibly because of the large epithelial surface
areas of the organ. Drugs have a relatively long residence time in the small
intestine, which also compensate for ionization effects.
3.7 According to the
pH-partition theory, absorption of a weak elec-trolyte drug depends on the
extent to which the drug exists in its unionized form at the absorption site.
According to the Henderson– Hasselbalch equation
pKa = pH + log( [HA]/[A−] )
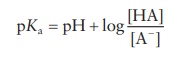
log
Cu/ Ci = p Ka –
pH
log
Cu/ Ci = 3 .5 − 2 = 1.5
where:
Cu is the concentration
of unionized drug
Ci is the concentration
of ionized drug
Cu / Ci
= antilog
1.5 = 31.62:1
In the plasma
pKa = pH
+ log [HA]/[A−]
log Cu/ Ci = p Ka
− pH
log Cu/ Ci = 3 .5 −
7 . 4 = −3.9
Cu/ Ci = antilog ( −
3 .9) = 0.00125
Therefore,
most of the administered aspirin remains unionized in the stomach and thus it
is rapidly taken up by the stomach, leading to gastric bleeding.
3.8 Six physicochemical
properties of a drug that influence drug absorp-tion are (1) molecular weight,
(2) drug solubility, (3) pKa,
(4) log P, (5) polymorphism, and (6)
stability. Physicochemical properties of a drug can be improved by salt
formation, bioconjugation, the use of cosolvents, and its use as a prodrug.
3.9
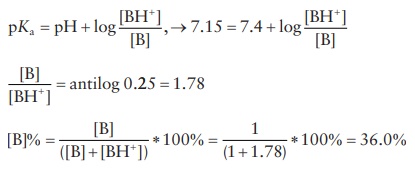
3.10 p Kb + pKa =
p K w , → p Ka =
pK w − p Kb,pKa
= 14. 0 − 5.6 = 8.4
At
pH 4.5,

3.11 pH = pKa +
log [salt]/[acid], →
5 .0 = 6 . 0 + log [salt]/[acid]
[acid] /[salt]= antilog(1.0) = 10:1
Related Topics