Factors affecting reaction kinetics
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Chemical kinetics and stability
Factors affecting reaction kinetics: 1. Temperature 2. Humidity 3. pH 4. Cosolvent and additives
Factors affecting
reaction kinetics
To
determine ways to prevent degradation of drugs in pharmaceutical for-mulations,
it is important to identify the mechanism of drug degradation and the factors
that affect the degradation rate or reaction kinetics. Once the route and
kinetics of degradation have been identified, stabilization strategies that
minimize reaction rates and maximize the shelf life of the drug product can be
adopted.
1. Temperature
If
a chemical reaction is endothermic (takes heat from the environment to react),
increase in temperature generally accelerates the reaction. If a reaction is
exothermic (gives out heat to the environment as it proceeds), temperature is
generally inversely proportional to reaction rate. Most chemical reactions of pharmaceutical
relevance in a dosage form are endothermic. Thus, an increase in temperature
generally accelerates the reaction rate.
Arrhenius equation
The
effect of temperature on the rate of drug degradation is expressed in terms of
the effect of temperature on the reaction rate constant, k, by the
Arrhenius equation (Figure 7.4):
k = Ae−Ea /RT (7.52)
where:
Ea is the activation
energy
A is a preexponential
constant
R is the gas constant
(1.987 calories/degree.mole)
T is the absolute
temperature (in Kelvin)
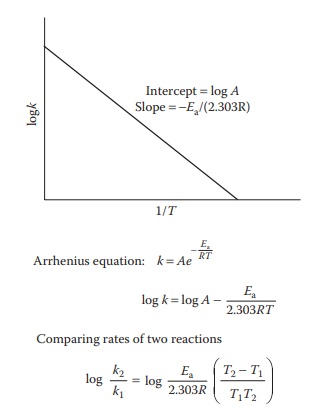
Figure 7.4 Arrhenius plot. Plot of the variation of the rate constant, k, versus
reciprocal of the absolute temperature, T.
The
Arrhenius expression can also be written as (Figure
7.4):
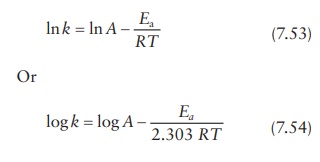
This
equation is of the form y = mx + c
for a straight-line plot. Thus, an Arrhenius plot of log k on the y-axis against
reciprocal of the absolute temperature (1/T)
on the x-axis yields Ea from the slope of the
straight line (Figure 7.4). This equation is
not amenable for direct application for the measurement of reaction rates,
since A and Ea are
unknown. Nevertheless, activation energy, Ea,
can be calculated by comparing reaction rates at two different temperatures.
Thus,
for temperatures T1 and T2,
k1 = Ae−Ea /RT1 (7.55)
k2 = Ae−Ea /RT2 (7.56)
Thus,
k2 / k1 = Ae−Ea/RT2 / Ae−Ea/RT1 = e Ea /RT1 − Ea/RT2 = eEa/R(1/ T1 −1/T2
) = eEa/R((T2 −T1)/ T1T2 )
Which
is same as:
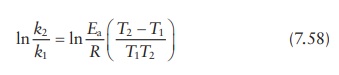
Or,
as in Figure 7.4,
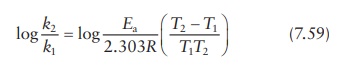
Thus,
measurement of the reaction rate constant, k,
at two different tempera-tures allows the calculation of the activation energy,
Ea, for a given reaction.
Shelf life
The
Arrhenius plot can be used to determine the shelf life of the drug. The
half-life (t1/2) and shelf
life (t0.90) expressions
from the reaction order can be substituted for the reaction rate constants, k, in the above equations to directly
infer product’s shelf life at a given temperature. These calcula-tions allow
the calculation of temperature of optimum drug stability over its shelf life.
If a drug is stable at room temperature (25°C), it is usually labeled for
storage at controlled room temperature (range 15°C–30°C). If a drug is unstable
at room temperature but stable at lower refriger-ated temperature (5°C), it is
usually labeled for storage under refrigerated conditions (range 2°C–8°C). This
is the case, for example, with various injectables such as penicillin, insulin,
oxytocin, and vasopressin. Typically, a shelf life of 24 months or more is
desired for all commercial products to allow enough time for manufacture,
storage, distribution, and con-sumption by the patient. Appropriate
temperature, packaging configura-tion, and drug product storage conditions are
determined to achieve the desired shelf life. Stabilization strategies for
drugs against degradation during storage are often required for achieving and
extending the desired shelf life.
Thermodynamics of reactions
Arrhenius
equation provides a mathematical basis of connecting reaction kinetics to the
collision theory and the transition state theory of chemical reactions. The
collision theory states that reactions happen by collisions that happen among
reacting molecules in a favorable configuration. It highlights the need for
several random collisions for effective collisions, which lead to the reaction,
to occur. The collision theory predicts an increase in intermolecular
collisions as a function of temperature, thus leading to higher number of
effective collisions and higher reactivity at higher temperatures.
The
transition state theory states that an activation energy barrier must be
surpassed for a reaction to become spontaneous. This activation energy barrier
can be understood as the energy of collisions required for them to overcome the
intermolecular repulsions at close contact for effective intermolecular
reactions to occur. Arrhenius equation relates the rate of a reaction, k, with the activation energy barrier, Ea.
Ink = In A – (Ea/RT)
Once
the activation energy barrier is overcome, the free energy difference between
the reactants and the products, ∆ G,
determines the reaction rate. This is given by the equation:
ΔG = −RT lnk (7.60)
In
addition, the free energy difference between the reactants and the prod-ucts is
a measure of the difference in the enthalpy, ∆H, and entropy, ∆S,
between the reactants and products. This is represented by the equation:
ΔG= ΔH−TΔS (7.61)
Greater
the free energy difference, that is, lower the free energy of the products than
that of the reactants, the faster the reaction. Thus, a negative ∆G facilitates a forward reaction. This
can be achieved by lower enthalpy of the products than that of the reactants,
achieving a negative ∆H, or higher
entropy of the products than the reactants, achieving a high ∆S and a negative T∆S.
These
equations can be used in conjunction with each other to connect a reaction’s
thermodynamic parameters to reaction rates, which can be deter-mined
experimentally. This allows the determination of thermodynamic parameters, such
as free energy and entropy change, of various reactions.
2. Humidity
Water
can influence reaction kinetics by acting as a reactant, a solvent (i.e., a
reaction medium), or a plasticizer.
Water as a reactant
For
hydrolytically sensitive drugs, water acts as a reactant and increases the drug degradation rate directly by
participating in a bimolecular reaction. Such reactions may follow second-order
or pseudo first-order kinetics, depending on whether water is available in the
reaction medium in lim-ited (such as contamination in a solvent or adsorbed
water in a solid-state excipient) or ample (such as the solvent) quantity.
Water as a plasticizer
A
plasticizer is a substance that is used as an additive to promote fluidity in a
solid state. For example, polyethylene glycol (PEG) is commonly used as a
plasticizer in tablet film-coating applications to allow the formation of a
flexible film that wraps around the tablet core. Small amounts of free water
(e.g., adsorbed on the surface) present in the solid particles can promote
local-ized dissolution and fluidity or flow of reacting molecular species.
Thus, for drugs that are not hydrolytically sensitive, water can increase
reaction rates by acting as a plasticizer
in the solid dosage forms, by increasing the molecular mobility and diffusion
rates of the reactive components. This is commonly seen in solid dosage forms
such as tablets and capsules, where the reaction rates are dependent on the
humidity during storage.
Water as a solvent
In
addition to the role of water as a solvent in the liquid dosage forms, small
amounts of adsorbed water can also act as a solvent
in the microen-vironment within a solid dosage form. This can affect reaction
rates by the following:
1. Solubilizing reacting components and increasing their
mobility
2. Affecting the disproportionation of the salt form of the
drug to its free acid or free base form, which may have different reactivity
compared with the salt form of the drug
3. Removing the product away from the reacting species, so
that equi-librium reactions proceed more rapidly toward the formation of the
product
Disproportionation
of the salt form of a drug in a solid dosage form to its constituent free acid
or free base form of the active pharmaceutical ingredi-ent (API) is commonly
attributed to the dissolution of the salt in the free water in the dosage form.
Determination and modeling the effect of water/humidity
Experimentally,
the effect of water or humidity on the stability of a dosage form is determined
by determining drug degradation kinetics at different temperature and humidity
storage conditions. This is accomplished by stor-ing the drug product under
open-dish conditions at different controlled temperature and humidity
conditions for different time periods, followed by analysis of the degradation
products. These studies are called isothermal
degradation rate studies, since the
temperature is kept constant throughout the
study.
The
effect of relative humidity (RH) at a
fixed temperature on drug’s degradation rate constant, k, can be incorporated using an empirically determined humidity
effect constant, B, as:
k = eB( RH) (7.62)
This
equation may be combined with Arrhenius equation for the effect of temperature
on reaction rate:
k = Ae -Ea/RT (7.63)
To
obtain,
k = AeB( RH )−Ea/RT (7.64)
This
combined equation predicts reaction rate as a function of both temperature and
humidity.
The
effect of humidity on reaction rate constant is an empirically fitted model.
Hence, this model can take different forms, depending on the experimental
system under investigation. For example, some systems may be better described
by the following equation:
k = AeEa / RT +B(RH) (7.65)
Nonetheless,
combining the humidity effect with the temperature effect on reaction rate
constant provides a better estimation of the extent of drug degradation over
its shelf life.
3. pH
Disproportionation effect
The
pH of the drug solution in a liquid dosage form and the microenviron-mental pH
in a solid dosage form can significantly influence drug stability by affecting
the proportion of ionized versus unionized species of a weakly acidic or a
weakly basic drug. The proportion of free acid or free base form of a drug at a
given pH is modeled by the Henderson–Hasselbalch
equation.
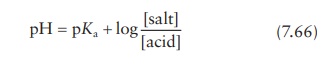
for
a drug that is the salt of a weak acid, or
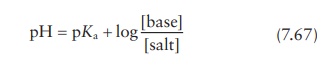
for
a drug that is the salt of a weak base.
Disproportionation
of a salt into its free acid or base form can influ-ence reactivity by changing
the concentration of the reacting species. Generally, the free acid or the free
base form of a drug is more reactive. Thus, drugs that are salts of free acids
are unionized in greater proportion and, consequently, are more reactive at
acidic pH, and drugs that are salts of free bases are unionized in greater
proportion and, consequently, are more reactive at basic pH.
Acid–base catalysis
Acid
(H+) and base (OH−) can
catalyze several reactions directly. For example, the rate of an ester
hydrolysis reaction catalyzed by hydrogen or hydroxyl ions can vary
considerably with pH. The H+ ion catalysis predomi-nates at a lower
pH and the OH− ion catalysis operates
at a higher pH.
Acids
and bases can affect reaction kinetics by specific
or general catalysis. For example, in specific catalysis, the reaction rate
depends only on the pH of the system and not on the concentration of actual
acid or base salts (such as HCl vs. HF or NaOH vs. KOH) contributing the H+
or the OH− ions. In general
catalysis, all species capable of donating or sequestering protons contribute
to the reaction rate, and proton transfer from an acid to the solvent or from
the solvent to a base is the rate-limiting step. General catalysis is usually
evident by changing reaction rates with changing buffer concentration at a
constant pH.
pH–rate profile
Rates
of chemical reactions are often determined at different pH values to identify
the pH of optimal drug stability. The pH–rate profiles are two-dimensional
plots of observed reaction rate constant (kobs)
on the y-axis against pH on the x-axis. The shape of a pH–rate profile
reflects on the mechanism of the reaction. For example, Figure 7.5 shows representative pH–rate profiles that indicate,
for the corresponding subfigures, (A) base catalysis and stability at acidic
pH, (B) acid catalysis and stability at basic pH, (C) a continuum of acid and
base catalysis with a narrow pH region of maximum drug stability, and (D) acid
and base catalysis under extreme ionization conditions and a wide pH region of
maximum drug stability.
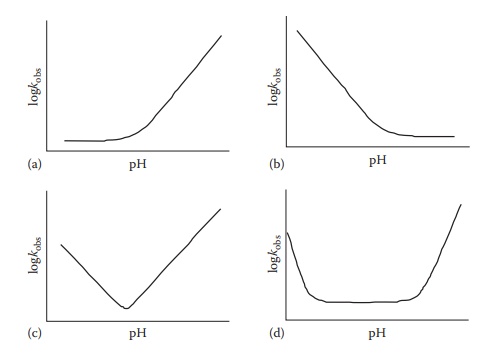
Figure 7.5 Typical pH stability profiles. Examples of pH—stability profiles for a
drug that degrades under basic (a), acidic (b), or both acidic and basic conditions
(c and d).
Proteins
are particularly sensitive to changes in pH, particularly with respect to the
conformation of the secondary and tertiary structures. Changes in the
ionization of amino acid side chains in proteins with changes in pH can lead to
folding or unfolding to varying degrees. Proteins exhibit overall charge
neutrality at their isoelectric point,
where the proportion of the positively charged groups within the protein is the
same as that of the negatively charged groups. Proteins tend to be most stable
in their most folded state, called the native state, which is generally
obtained by appro-priate balance of charged and uncharged groups on the
surface. The pH of optimal stability can be determined by plotting log k against pH. For example, recombinant α-antitrypsin (rAAT) has a V-shaped
pH—stability profile, with optimal stability at pH 7.5.
4. Cosolvent and additives
For
liquid dosage forms, cosolvents are frequently used to improve drug solubility
and stability in the vehicle. These cosolvents are commonly one or more of PEG, propyleneglycol (PG),
and ethanol. In addition, water-miscible surfactants, such as polysorbate
80, and polymers, such as polyvinyl alco-hol (PVA), may be used. Other common
components of liquid dosage forms include buffers to maintain desired pH, ionic
components for isotonicity of parenteral solutions, preservatives, sweeteners,
flavors, and colorants.
These
additives in liquid formulations lead to simultaneous changes in
physicochemical conditions of the reaction medium, such as dielectric con-stant, ionic strength, surface tension, and viscosity,
all of which may affect rates of
chemical reactions. The effect of ionic strength and dielectric con-stant
depends on the relative hydrophilicity of the reactants and products. If, for example,
products achieve greater solubilization in the reaction medium, the rate of the
reaction would be higher. This is due to the ability of the products to diffuse
away from the reaction site, leading to shift in the equi-librium of the
reaction toward the formation of the products. Similarly, if the reacting
species have opposite charges, a solvent with a low dielectric constant
accelerates the reaction rate. This could be attributed to lower
sol-ubilization of the reacting species, which also have affinity with each
other, thus increasing the propensity for the reaction. On the other hand, if
the reacting species have the same charge, a solvent with high dielectric
constant will accelerate the reaction by forming bonds and dissolving both
species, thus reducing intermolecular repulsion due to like charges.
Drug–excipient interactions
Chemical
interaction between components in solid dosage forms can impact, often
increasing, the rate of drug degradation.
Buffer salts are often added to
maintain a formulation at optimal pH. These
salts may often affect the degradation rate. For example, the hydrolysis rate
of codeine is almost 20 times higher in phosphate buffer at neutral pH than in
an unbuffered solution at this pH. At neutral pH, the major buffer species are
H2PO4−
and HPO42−,
either of which may act as a general base catalyst for codeine degradation.
Excipients
that have specific functional groups such as the carboxylate group on
croscarmellose sodium or the sulfate group on sodium lauryl sulfate can exhibit
specific interactions with the drug substance that can destabilize a drug.
These interactions can be direct reaction, salt disproportionation, or
facilitation of a reaction by surface adsorption. In addition, excipients often
contain small quantities of reactive substances, called reactive impurities.
These reactive impurities in excipients can react with low-dose drugs in the
dosage form to cause drug degradation. For example, PEG and
polyvinyl-pyrrolidone (PVP) commonly have peroxide impurities that can cause
oxi-dative degradation of sensitive drugs.
The
effect of excipients on drug stability is usually assessed early in drug
development through excipient
compatibility studies. Drug degradation rate is determined in physical
mixtures of a drug with individual or a combina-tion of excipients. Excipient
compatibility studies are also useful later in drug development when unexpected
impurities are observed. Mechanistic investigation of the reaction pathway
leading to the formation of these impurities becomes a cornerstone of drug
product stabilization strategies.
Catalysis
Components of a dosage form can frequently act as, or bring in species that act as, reaction catalysts. A catalyst affects the rate of change in the concentra-tions of products and reactants in a chemical reaction but not the equilibrium concentration of reactants and products in the reaction. As seen in Figure 7.6, a catalyst may change the reaction pathway and lower the energy of activation required for a reaction, thus accelerating the reaction.
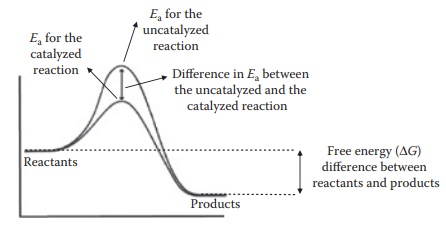
Figure 7.6 Effect of catalyst. Transition state during reaction progress (on the
x-axis from left to right) with the energetics (on the y-axis) is indicated by
the peak in the energy requirement for the reactants to convert to products.
The presence of a catalyst changes the reaction pathway such that the height of
this peak is lowered.
However, the
thermodynamic driver for a reaction, that is, free energy difference between
the reactants and the products, remains the same for an uncata-lyzed versus a
catalyzed reaction. Thus, a catalyst
influences the speed but not the
extent of a reaction. In addition, a catalyst does not get chemically altered itself.
In
pharmaceutical dosage forms, heavy metal contaminants in excipients and drug
substances often act as catalysts.
Related Topics
