Protein binding
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Complexation and protein binding
A molecule (drug) that binds the protein is known as a ligand, and the protein with which it binds is called the substrate.
Protein binding
A
molecule (drug) that binds the protein is known as a ligand, and the protein
with which it binds is called the substrate.
Protein
binding is involved in the following:
·
Plasma protein binding of drugs in the central or plasma
pharmacoki-netic compartment after administration.
·
Drug–receptor interactions (when the receptor is a protein)
leading to drug action.
·
Substrate–enzyme interactions leading to enzyme action or
inhibition.
Physical
parameters of protein–ligand binding interaction include the kinet-ics of
binding and its thermodynamics.
Kinetics of ligand–protein binding
Binding
of a ligand (L) to a protein (P) to form a protein–ligand complex (PL) can be
expressed as:
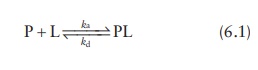
where,
ka and kd are the equilibrium rate
constants known as the associa-tion constant and the dissociation constant,
respectively.
Their
rate expressions can be written as:

The
dissociation constant (kd)
has a unit of concentration (such as M), while the association constant (ka) has the unit of inverse
concentration (such as M−1).
Thus,
kd
= 1/ka (6.4)
1. Parameters of interest
Biopharmaceutical
applications of protein binding require the determina-tion of two key
parameters:
1. Binding affinity (defined as the association constant, ka)
2. Binding capacity (maximum number of ligand molecules that
can be bound per molecule of protein, ymax)
2. Experimental setup
Protein–ligand
binding studies are usually carried out with fixed protein concentration and
varying ligand concentration, or vice versa. At each con-centration, the amount
of ligand bound is separated from free ligand by techniques such as
centrifugation and filtration. Free ligand concentra-tion is then determined by
analytical methods such as ultraviolet-visible spectroscopy (UV-VIS). The
measurement of free ligand concentration as a function of total ligand
concentration enables the determination of both the affinity and the capacity
of ligand binding of the substrate.
The
amount of ligand bound to the substrate in each experiment (y) can be expressed as a fraction of
maximum concentration that can be bound (ymax),
as
Θ = y / ymax (6.5)
Where,
y represents the molar concentration, the amount of ligand bound per unit molar
concentration, or the amount of protein, and ymax represents the maximum binding capacity.
3. Determining ka and ymax
Nonlinear regression I
Average
number of ligand molecules bound per molecule of protein is expressed as the
molar concentration of ligand bound to the protein per molar concentration of
the protein. For the case of single binding site on the protein, molar
concentration of ligand bound to the protein is given by [PL] and the total
protein concentration is given by [P] + [PL]. Thus,
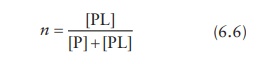
From
the expression for the dissociation constant, kd,
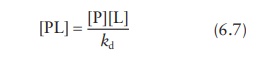
Combining
these two equations,
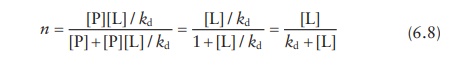
For
a single ligand-binding site per protein,
n = θ (6.9)
Thus, the amount of ligand bound to the protein as a fraction of sat-uration concentration (θ), which is experimentally determined, can be written as:

Directly
plotting θ against the free
ligand concentration [L] gives a satura-tion curve (Figure
6.8a), and the data can be fitted by nonlinear regression to solve for ymax and kd as parameters.
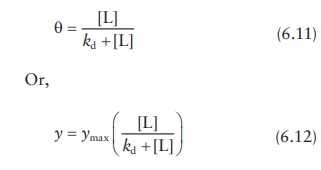
However,
nonlinear regression is a computationally intensive parameter-estimation method
that uses algorithms for adjusting the equation parameters to best fit the
data. Thus, it suffers the drawbacks of requiring software support, being
dependent on the initial values of parameters chosen, and the possibility of
coming up with incorrect parameters due to minimization of sum-of-square errors
in a local region. Therefore, lineariza-tion of this equation followed by
simple linear regression is traditionally preferred. Two methods for
linearization are the double-reciprocal plot and the Scatchard plot.
Linear regression I: Double-reciprocal (Hughes–Klotz) plot
Inverting
Equation 6.11,
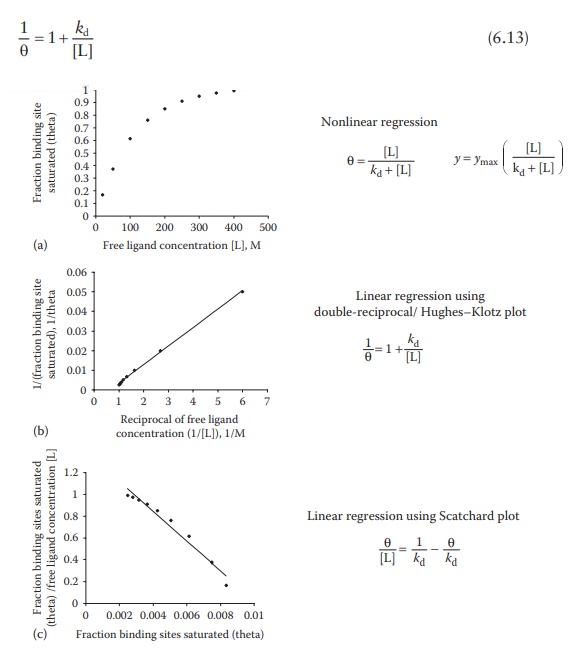
Figure 6.8 Methods for determining
ligand–protein interaction parameters.
In
this equation, [L] represents the free ligand concentration, which is also
experimentally determined. This is a linear form of the equation, whereby
plotting 1/θ against 1/[L] gives
a straight line, with slope as kd.
This plot is known as the double-reciprocal plot, Lineweaver– Burk plot,
Benesi–Hildebrand binding curve, or the Hughes–Klotz plot (Figure 6.8b).
As
seen in Figure 6.8b, graphical treatment of
data using Klotz reciprocal plot heavily weighs those experimental points
obtained at low concentrations of free ligand and may, therefore, lead to
misinter-pretations regarding the protein-binding behavior at high
concentrations of free ligand. The Scatchard plot (Figure
6.8c)—discussed in the next section—does not have this disadvantage and
is, therefore, preferred for plotting data.
Linear regression II: Scatchard plot
The
equation for θ can also be
converted into:

Adding
and subtracting 1/kd from
this equation:
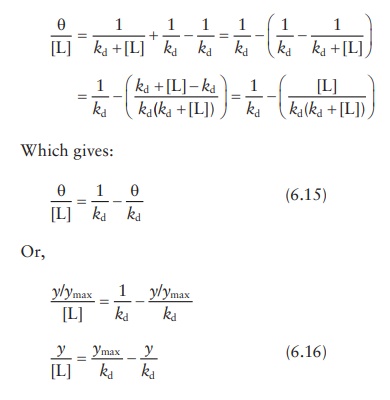
Thus,
given that both θ and [L] are
experimentally determined, plotting θ/[L] against θ would give a slope
of −1/kd and an intercept of ymax/kd. This linear plot is known as the
Scatchard plot (Figure 6.8c). Interchanging the
x- and y-axis of the Scatchard plot results in the Eadie–Hofstee plot.
Although
the Scatchard plot is widely used for protein–ligand bind-ing data analyses, it
suffers from mathematical limitations. As seen in Figure
6.8c, the Scatchard transformation distorts
experimental error, resulting in violation of the underlying assumptions of
linear regression, viz., Gaussian distribution of error and standard deviations
being the same for every value of the known variable. In addition, plotting θ/[L]
against θ leads to the unknown variables being a part of both x- and y-axis, while linear regression assumes that y-axis is unknown and x-axis
is precisely known.
Thermodynamics of ligand–protein binding
Binding
affinity can also be inferred from the thermodynamics of binding. A binding
interaction, where more stable bonds are formed than are broken, involves
release of energy as heat. The amount of heat released can be pre-cisely
measured in carefully controlled experiments by a technique generally known as
calorimetry. For example, isothermal titration calorimetry (ITC) involves the
titration of one binding partner (ligand) into another (protein) while
measuring the heat (enthalpy) change per unit volume of the ligand added to the
protein. These data are integrated to yield enthalpy change per mole of the
injectant and plotted against the molar ratio of ligand to protein (Figure 6.9). In this plot, the enthalpy difference
between the starting value and the saturated value indicates enthalpy (∆H) of binding, the slope of the
transition indicates binding affinity, and the ligand/protein molar ratio at
the inflexion point indicates the stoichiometry of binding, that is, the num-ber
of ligand molecules binding per protein molecule.
Thus,
ITC can be used to determine the thermodynamic parameters associated with a
physical or a chemical change. These parameters include the free energy (∆ G), enthalpy (∆ H), and entropy (∆ S)
change, which are related to each other as:
∆G= ∆H−T∆S
Spontaneous
processes must have favorable overall free energy of reaction (negative ∆ G). The ITC helps determine whether the
ligand–protein binding is enthalpically driven (negative ∆ H) or entropically driven (positive ∆ S).
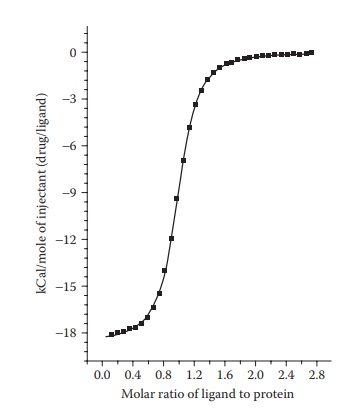
Figure 6.9 A typical ITC
thermogram.
An
entropically driven process is likely to be significantly influenced by the
liquid medium. In the case of an enthalpically driven process, the binding
constant and the enthalpy change associated with the binding are indicative of
the strength of binding. Complexation is a binding process whereby the degrees
of freedom of two or more molecules are reduced as they bind each other. Thus,
complexation is an entropically unfavorable process (i.e., has a negative
entropy).
An
ITC experiment can also help determine the dissociation constant (kd).
ΔG = −RT lnkd (6.17)
where:
R is the gas constant
T is the absolute
temperature
Factors influencing protein binding
The
physicochemical characteristics and concentration of the drug, the pro-tein,
and the characteristics of the liquid medium in which binding takes place
influence drug (ligand)–protein binding.
1. Physicochemical characteristics and concentration of the drug
The
extent of protein binding of many drugs is a linear function of their oil–water
partition coefficient (Figure 6.10), which is a
measure of their hydrophobicity. Thus, protein binding generally increases with
an increase in drug lipophilicity. This indicates involvement of drug– protein
hydrophobic interactions. This phenomenon can be used to predict the biological
activity of a drug’s analogs. For example, an increase in the
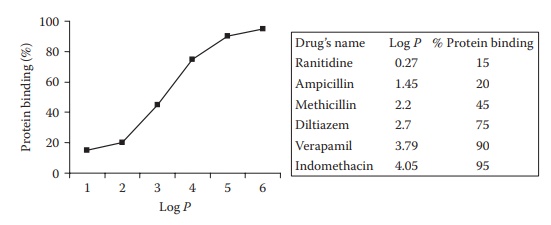
Figure 6.10
Effect of lipophilicity (log P) on plasma protein binding of drugs.
lipophilicity
of penicillins results in decreased activity. The hydrophobic binding of
penicillin in serum proteins reduces their potency in vivo, by decreasing their free plasma concentration.
Increasing
the concentration of the drug would generally increase the extent of binding.
However, if the concentration is increased beyond the saturation concentration,
saturation of some or all binding sites can occur and the proportion of drug
bound would actually decrease, as the absolute amount of bound drug remains
constant.
2. Physicochemical characteristics and concentration of the protein
In
a dilute solution, increasing protein concentration is expected to increase the
proportion of the drug bound. However, at high protein concentrations, the
protein may agglomerate or self-associate, leading to shielding of the
hydrophobic region(s), which can reduce drug binding if the drug—protein
interaction is driven by hydrophobic interactions.
Physicochemical
characteristics of the protein, such as the density dis-tribution of
hydrophobic groups on its surface, significantly influence the extent of drug–protein
interaction. Thus, binding affinity of a drug toward different proteins can be
markedly different.
3. Physicochemical characteristics of the medium
Binding
interaction between the drug and the protein involves disruption of
drug–solvent and protein–solvent bonds with the formation of drug– protein
bonds. Thus, solvent medium that strongly interacts with either or both of the
drug and protein can lead to thermodynamically unfavorable outcome of
drug–protein interactions. In addition, change in the dielectric constant of
the medium, such as in the presence of alcohol in aqueous solutions, can lead
to altered forces of attraction and bonding between the drug and the protein.
For example, salt concentration and dielectric constant of the solvent medium
can significantly influence drug–protein interactions.
Plasma protein binding
Systemically
administered drugs reach target organs and tissues through blood, which is a
mixture of several substances, including proteins. In pharmacokinetic terms,
the blood or the plasma is called the central com-partment. In this
compartment, drugs often bind plasma proteins. The drug exits the central
compartment as it partitions into organs and tissues, called the peripheral
compartment. Plasma protein binding of drugs is generally reversible, so that
protein-bound drug molecules are released as the level of free drug in blood
declines.
1. Plasma proteins involved in binding
Blood
plasma normally contains about 6.72 g of protein per 100 cm3, the
protein comprising 4.0 g of albumin, 2.3 g globulin, and 0.24 g of fibrinogen.
Albumin (commonly called human serum albumin [HSA]) is the most abundant
protein in plasma and interstitial fluid. Plasma albumin is a globular protein
consisting of a single polypeptide chain of molec-ular weight 67 kDa. It has an
isoelectric point of 4.9 and, therefore, a net negative charge at pH 7.4.
Nevertheless, albumin is amphoteric and capable of binding both acidic and
basic drugs. Physiologically, it binds relatively insoluble endogenous compounds,
including unesterified fatty acids, bilirubin, and bile acids. Human serum
albumin has two sites for drug binding:
1. Site I (warfarin site) binds bilirubin, phenytoin, and
warfarin.
2. Site II (diazepam site) binds benzodiazepines,
probenecid, and ibuprofen.
Plasma
proteins other than albumin are sometimes the major binding part-ners of drugs.
For example, dicoumarol is bound to β- and α-globulins, and certain steroid
hormones are specifically and preferentially bound to particular globulin
fractions. Among other proteins, α1-acid glycoprotein (AAG) binds to
lipophilic cations, including promethazine, amitriptyline, and dipyridamole.
2. Factors affecting plasma–protein binding
The
amount of a drug that is bound to plasma proteins depends on three factors:
1. Concentration of free drug
2. Drug’s affinity for the protein-binding sites
3. Concentration of protein
3. Consequences of plasma–protein binding
The
binding of drugs to plasma proteins can influence their action in a number of
ways:
1. Reduce free drug concentration. Protein binding affects
antibiotic effectiveness, as only the free antibiotic has antibacterial
activity. For example, penicillin and cephalosporins bind reversibly to
albumin, thus affecting their free concentrations in plasma.
2.
Reduce drug diffusion. The bound drug assumes the diffusional and other
transport characteristics of the protein molecules.
3. Reduce volume of distribution. Only free drug is able to
cross the pores of the capillary endothelium. Protein binding will affect drug transport
into other tissues. When binding occurs with high affinity, the drug is
preferentially localized in the plasma or the central com-partment. In
pharmacokinetic measurements, this reflects as a low volume of distribution of
the drug.
However,
some drugs (e.g., warfarin and tricyclic antidepressants) may exhibit both a
high degree of PPP and a large volume of distri-bution. Although drug bound to
plasma proteins is not able to cross biological membranes, binding of drugs to
plasma proteins is in a dynamic equilibrium with the drug bound to plasma
proteins. If the unbound (or free) drug is able to cross biological membranes
and has a greater affinity and capacity for binding to the tissue biomolecules,
compared with the plasma proteins, the drug may exhibit high vol-ume of
distribution, despite also exhibiting high PPP. As free drug moves across
membranes and out of vascular space, the equilibrium shifts, drawing drug off
the plasma protein to replenish the
free drug lost from vascular space. This free drug is now also able to traverse
membranes and leave vascular space. In this way, a drug with a very low free
fraction (i.e., a high degree of PPP) can exhibit a large volume of
distribution.
4. Reduce elimination. Protein binding retards the
metabolism and renal excretion of the drug. Proteins are not filtered through
glomerular filtration. Thus, protein-bound drugs have reduced rate of
filtration in the kidneys and metabolism in the liver.
5. Increase risk of fluctuation in plasma free drug
concentration.
a. In cases where a drug is highly protein-bound (around
90%), small changes in binding, protein concentration, or displacement of the
drug by another coadministered drug (drug–drug interaction) can lead to drastic
changes in the concentration of free drug in the body, thus affecting efficacy
and/or toxicity.
However,
a plasma protein may have multiple binding sites. Thus, if drugs bind to
different sites on a protein, there will not be a competitive binding
interaction between them. Thus, some drugs that are highly bound to albumin
exhibit competitive inter-actions, while others do not.
b. Sometimes, drug administration may also cause
displacement of body hormones that are physiologically bound to the protein,
thus increasing free hormone concentration in the blood.
c.
Disease states that alter plasma protein concentration may alter the protein
binding of drugs. If the concentration of protein in plasma is reduced, there
may be an increase in the free fraction of the drugs bound to that protein.
Similarly, if pathological changes in binding proteins reduce the affinity of
drug for the protein, there will be an increase in the free fraction of drug.
Effect on dosing regimen
Plasma
protein binding can affect dosing regimen of a drug in several ways.
1. Lower metabolism and elimination of a
plasma-protein-bound drug can lead to longer plasma half-life, compared with an
unbound drug. Thus, the protein-bound drug may serve as a reservoir of drug
within the body, maintaining free drug concentration through equilibrium
dissociation process. This leads to long half-life and sustained plasma
concentrations. Thus, dosing frequency would need to be adjusted in cases where
drug’s PPP or the concentration of plasma proteins is affected (such as burns).
2. Dose adjustments are frequently required in the case of
disease states that affect the protein to which the administered drug is bound.
Certain disease states increase AAG concentration while reducing albumin
concentration. For example, acute burns reduce the con-centration of
circulating albumin, resulting in an increase in the free fraction of drugs
that are normally bound to albumin. On the other hand, AAG concentration is
substantially increased after an acute burn, resulting in a decrease in the
free fraction of drugs that are normally bound to this plasma protein.
3.
Age-based dose adjustments often have to account for PPP of drugs. For example,
newborns have selectively lower plasma protein levels than adults. Thus,
although the neonatal HSA concentration at birth is 75%–80% of adult levels,
AAG concentration is only ~50%. Thus, dose adjustment may be needed for drugs
that bind AAG.
4.
Drug–drug interactions. Drugs that compete for the same plasma-protein-binding
site can displace one another. This can lead to increased free level of a drug.
Minor perturbation in PPP can have a significant influence on free drug
concentration. Thus, coadministration of cer-tain drugs may be contraindicated
or require dose adjustment.
Drug receptor binding
Target
protein (receptor) binding is routinely utilized in drug discovery, with the
goal of maximizing binding affinity and specificity. This is expected to result
in a drug molecule that is highly potent and has low off-target activ-ity and,
thus, toxicity. The principles involved in delineating the kinetics of
drug–receptor binding are same as discussed earlier for ligand–protein binding.
Substrate enzyme binding
Binding
of a ligand, which serves as a substrate for an enzyme, to an enzyme is a part
of a continuous process involving conversion of the substrate into the
product(s) by the enzyme. This process involves continuous recycling of the
enyzme’s binding sites for fresh substrate, as each molecule of the substrate
is converted into product(s). Thus, substrate–enzyme binding kinetics are
represented in terms of the rate of binding, and the satura-tion of the binding
kinetics is considered in terms of the maximum rate of binding.
The
rate of binding kinetics for substrate–enzyme reactions follows a hyperbolic
function, described by the Michaelis–Menten equation.

where:
v is the initial
reaction rate
vmax is the maximum reaction rate
[S]
is the substrate concentration
kM is the Michaelis–Menten constant, which represents the ratio
of the rate of dissociation of the
enzyme–substrate complex to its rate of formation.
The
similarity of this equation to Equation 6.7
indicates similar basic prin-ciples involved in their derivation.
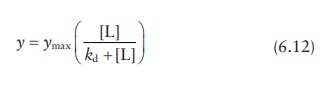
Related Topics
