Mechanisms of Renal Adverse Drug Reactions
| Home | | Pharmacovigilance |Chapter: Pharmacovigilance: Renal Adverse Drug Reactions
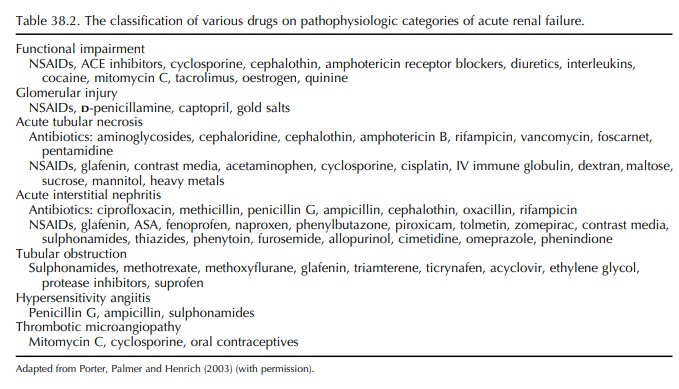
Drugs may adversely affect renal function by induc-ing structural injury to components of the nephron and/or by interfering with the filtration and transport processes or regulatory pathways.
MECHANISMS OF RENAL ADVERSE DRUG
REACTIONS
Drugs
may adversely affect renal function by induc-ing structural injury to
components of the nephron and/or by interfering with the filtration and
transport processes or regulatory pathways (Table 38.2).
Drugs
interfering with glomerular blood flow may induce functional renal impairment.
Cyclosporine and epinephrine cause preglomerular arteriolar vasocon-striction
resulting in a decrease in intra-glomerular pressure and filtration pressure.
In clinical condi-tions in which systemic vasoconstriction is promi-nent like
dehydration or heart failure, glomerular blood flow is critically dependent
from a counteract-ing vasodilation of the preglomerular arteriole medi-ated by
compensatory PGE2 and PGI2 production (Whelton, 1999). In the same patients,
maintenance of adequate glomerular filtration pressure is also depen-dent of
postglomerular vasoconstriction mediated by angiotensine II. Disruption of
these counter-regulatory mechanisms by the administration of NSAIDs or of drugs
interfering with angiotensine II (ACE inhibitors and angiotensine II receptor
blockers) can produce clinically important and even severe deterioration in
renal function. When NSAIDs and ACE inhibitors are co-prescribed there is an
accrued risk for functional renal impairment. This drug combination should be
avoided, especially in elderly patients and those taking diuretics (Adhiyaman et al., 2001).
The
publication of the Randomized Aldactone Eval-uation Study (RALES) (Pitt et al., 1999) promoted the combined use
of the anti-aldosterone agent spirono-lactone and ACE inhibitors in heart
failure patients. In the setting of this randomised clinical trial, the
incidence of severe hyperkalaemia was minimal, patients with renal failure or
pre-existing hyper-kalaemia being excluded from the trial. In subse-quent
years, however, case reports of life-threatening hyperkalaemia in patients
treated with spironolactone appeared in the literature (Schepkens et al., 2001). It became evident that
hyperkalaemia is episodic in these patients and linked to ARF. The main causes
for ARF in this setting were dehydration and wors-ening heart failure. In a
population-based time-series analysis recently conducted in Canada, an increase
was found in hyperkalaemia-associated morbidity and mortality in elderly
patients after abrupt increases in the prescription rate for spironolactone
following the publication of RALES (Juurlink et al., 2004).
Drug-induced
immune nephropathies include glomerulopathies and tubulointerstitial nephritis.
NSAIDs are known to induce both types of renal injury. A review of
NSAID-induced nephropathy reported an incidence of 39.2% of minimal change
glomerulopathy, 19.6% of tubulointerstitial nephri-tis, 13.4% of focal
glomerular sclerosis and 8.2% of other types of nephropathy (Ravnskov, 1999).
Gold salts previously used in rheumatoid arthritis induce a membranous
glomerulopathy. The disease is related neither to dose nor to the duration of
treatment, but susceptible seemed to be genetically controlled, HLA DR3-positive
patients being more prone to develop this adverse reaction. Drug-induced
interstitial nephri-tis represents a minority of ARF cases. Clinically, the
disease is characterised by bilateral lumbar pain, fever and skin rash. Many
patients exhibit hyper-eosinophylia, hypereosinophyluria and increased IgE
serum levels. In renal biopsy the characteristic lesions are interstitial
mononuclear cell infiltrates and tubular cell injury. Most often renal function
recovers after withdrawal of the drug with or without concomitant steroid
therapy. The drugs that are most frequently responsible for tubulointerstitial
nephritis are anti-biotics, mainly -lactams, and NSAIDs.
The
particular susceptibility of the tubular cell to nephrotoxic injury has several
reasons. Tubular solute transport and other renal metabolic processes utilise
considerable oxygen and are susceptible to the action of metabolic inhibitors.
It is worthwhile to note that the S3-segment of the proximal tubule has the
highest rate of oxygen consumption per gram of tissue of the whole body.
Moreover, the renal tubular epithelium is the only place where protein-bound
drugs dissociate, traverse the renal epithe-lium and either accumulate in the
proximal tubular cell or reach the tubular lumen. An abundance of tubular
enzymes involved in tubular transport may be blocked, in view of the high
urinary to plasma concentration ratios exceeding 1000 in some cases. Typical
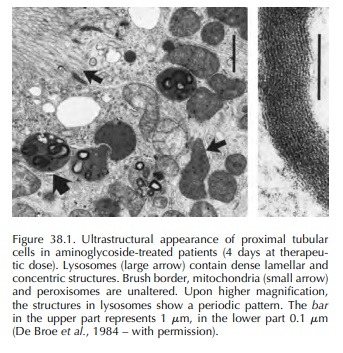
tubulotoxic drugs that are extensively stud-ied are the aminoglycoside
antibiotics (Verpooten, Tulkens and Molitoris, 2003). Aminoglycosides are polar
drugs that are freely filtered via the glomeru-lar membrane. Following binding
to megalin in the proximal tubular brush border, aminoglycosides traf-fic via
the endocytic system to lysosomes, where they accumulate in large amounts. In
lysosomes, aminogly-cosides induce an intense phospholipidosis by inhibit-ing
phospholipidases A and C and sphingomyelinase. This phospholipidosis occurs
rapidly involving all major phospholipids and is responsible for the forma-tion
of the so-called ‘myeloid bodies’ (Figure 38.1). At present it is unknown
whether phospholipidosis is linked to tubular cell necrosis. Besides
lyso-somes, aminoglycoside-induced alterations of mito-chondria have also been
described. More recently, proteomic analysis following gentamicin
administra-tion indicated energy production impairment and a mitochondrial
dysfunction occurring in parallel with the onset of nephrotoxicity (Charlwood et al., 2002). The severity of
aminoglycoside nephrotoxicity can be dissociated from the height of the peak of
the amino-glycoside blood level. It became evident that for a given total daily
dose the toxicity was greatest when the daily dose was being divided into
multiple small administrations. The reason for this apparent paradox is that
the renal cortical drug uptake is saturable, so that maintaining a low blood
level maximises tubular cellular drug uptake (Verpooten et al., 1989).
In
the distal part of the nephron, urine is concen-trated, and the likelihood of
crystalline precipitation increases substantially. ARF may result from tubular
obstruction due to intratubular precipitation of the drug or its metabolite.
This mechanism has been incriminated in the clinical syndrome of bilateral
flank pain and ARF associated with the use of suprofen (Henann and Morales,
1986; Hart, Ward and Lifs-chitz, 1987). This renal adverse drug reaction led to
the withdrawal of this NSAID from the market in 1986 (Chapter 1 of this book).
Because suprofen is a uricosuric agent, one might speculate that it could lead
to intratubular or ureteral precipitation of uric acid (Abraham et al., 1988). More recently, there have
been reports of this type of renal adverse event following high-dose
intravenous acyclovir and during treatment with protease inhibitors.
The
immunosuppressive drug cyclosporine is of particular interest since it can
display all types of nephrotoxicity (reviewed in Bosmans and De Broe, 2006). Cyclosporine
profoundly alters renal and glomerular haemodynamics. Administra-tion of
cyclosporine induces a decline in glomeru-lar filtration rate (GFR) and renal
blood flow by vasoconstriction at the level of the afferent arte-rioles.
Catecholamines, endothelin and eicosanoids like thromboxane are potential
mediators of this effect. Effects of cyclosporine on tubular func-tion consist
of increased proximal reabsorption of sodium resulting in decreased distal
sodium deliv-ery interfering with the potassium secretory capacity of the
distal tubule. This pathophysiologic effect may explain the observed
hyperkalaemic metabolic acidosis in cyclosporine-treated kidney allograft
recipients. Besides these functional side effects, cyclosporine induces
morphologic alterations in the kidney. First, cyclosporine induces
dose-dependent acute tubular changes consisting of isometric vacuoli-sation of
tubular cells, accumulation of eosinophilic bodies representing giant
mitochondria and micro-calcifications in proximal tubules. These patho-logic
alterations are reversible after dose reduction or withdrawal of cyclosporine.
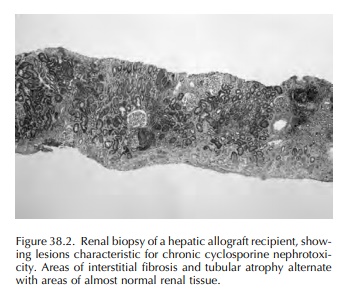
In contrast to the acute injury, chronic administration of cyclosporine may
lead to irreversible histopathologic lesions. They include renal arteriolar damage
(the so-called cyclosporine associated arteriolopathy), tubular atro-phy and
focal or striped interstitial fibrosis as well as glomerular sclerosis (Figure
38.2). Clinically, chronic cyclosporine nephrotoxicity is associated with
hypertension, progressive renal failure and a vari-able degree of proteinuria.
Thrombotic microangiopa-thy is an uncommon but serious adverse effect of
cyclosporine. The striking morphologic changes, resembling haemolytic-uraemic
syndrome, are exten-sive thrombotic processes in the renal microcircu-lation,
with several glomerular capillaries occluded by thrombi extending from the
afferent arterioles (Verpooten et al.,
1987). Laboratory findings include thrombocytopenia, haemolytic anaemia and
deterio-rating renal function.
Related Topics
