One-Compartment Open Model : Intravenous Bolus Administration
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Compartment Modelling
When a drug that distributes rapidly in the body is given in the form of a rapid intravenous injection (i.e. i.v. bolus or slug), it takes about one to three minutes for complete circulation and therefore the rate of absorption is neglected in calculations.
One-Compartment Open Model
Intravenous Bolus Administration
When a drug that distributes rapidly in the body is
given in the form of a rapid intravenous injection (i.e. i.v. bolus or slug),
it takes about one to three minutes for complete circulation and therefore the
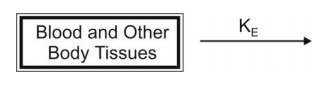
rate of absorption is neglected in calculations. The model can be depicted as
follows:
The general expression for rate of drug presentation to the body is:
dX/dt = Rate in (availability) - Rate out
(elimination) (9.1)
Since rate
in or absorption is absent, the equation becomes:
dX / dt = - Rate out (9.2)
If the rate
out or elimination follows first-order kinetics, then:
dX/dt = KΕ X (9.3)
where, KE = first-order elimination rate
constant, and
X = amount of drug in the body at any time t
remaining to be eliminated.
Negative sign indicates that the drug is being lost
from the body.
Estimation of Pharmacokinetic Parameters
For a drug that follows one-compartment kinetics
and administered as rapid i.v. injection, the decline in plasma drug
concentration is only due to elimination of drug from the body (and not due to
distribution), the phase being called as elimination phase. Elimination phase can be characterized
by 3 parameters—
1. Elimination rate constant
2. Elimination half-life
3. Clearance.
Elimination Rate Constant:
Integration of equation 9.3 yields:
ln X = ln Xo – KE t (9.4)
where, Xo = amount of drug at time t =
zero i.e. the initial amount of drug injected.
Equation 9.4 can also be written in the exponential
form as:
X = Xo e–KEt (9.5)
The above equation shows that disposition of a drug that follows one-compartment kinetics is monoexponential.
Transforming equation 9.4 into common logarithms
(log base 10), we get:
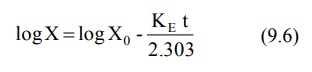
Since it is difficult to determine directly the
amount of drug in the body X, advantage is taken of the fact that a constant
relationship exists between drug concentration in plasma C (easily measurable)
and X; thus:
X = Vd C (9.7)
where, Vd = proportionality constant
popularly known as the apparent volume of
distribution. It is a pharmacokinetic parameter that permits the use of
plasma drug concentration in place of amount of drug in the body. The equation
9.6 therefore becomes:
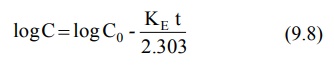
where, Co = plasma drug concentration
immediately after i.v. injection.
Equation 9.8 is that of a straight line and
indicates that a semilogarithmic plot of log C versus t will be linear with Y-intercept log Co. The
elimination rate constant is directly obtained from the slope of the line (Fig.
9.2b). It has units of min–1.
Thus, a linear plot is easier to handle mathematically than a curve which in
this case will be obtained from a plot of C versus t on regular (Cartesian)
graph paper (Fig. 9.2a).
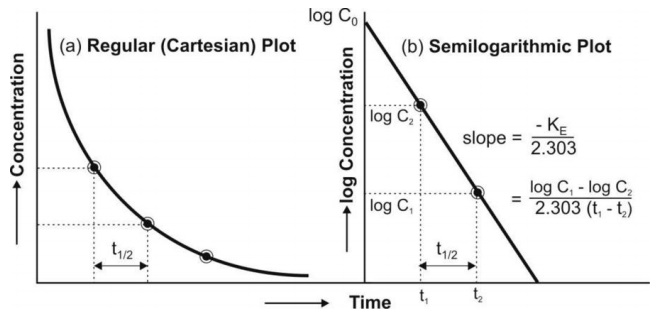
Fig. 9.2 (a) Cartesian plot of a drug that follows one-compartment kinetics and
given by rapid i.v. injection, and
(b) Semilogarithmic plot for the rate of elimination in a one-compartment
model.
Thus, Co, KE (and t½)
can be readily obtained from log C versus t graph. The elimination or removal
of the drug from the body is the sum of urinary excretion, metabolism, biliary
excretion, pulmonary excretion, and other mechanisms involved therein. Thus, KE
is an additive property of rate constants for each of these processes and
better called as overall elimination rate constant.
KE = Ke + Km + Kb
+ Kl + ...... (9.9)
The fraction of drug eliminated by a particular
route can be evaluated if the number of rate constants involved and their
values are known. For example, if a drug is eliminated by urinary excretion and
metabolism only, then, the fraction of drug excreted unchanged in urine Fe
and fraction of drug metabolized Fm can be given as:

Elimination Half-Life: Also
called as biological half-life, it
is the oldest and the best known of
all pharmacokinetic parameters and was once considered as the most important
characteristic of a drug. It is defined
as the time taken for the amount of drug in the body as well as plasma concentration to decline by
one-half or 50% its initial value. It is expressed in hours or minutes. Half-life is related to elimination rate
constant by the following equation:
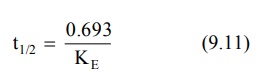
Elimination half-life can be readily obtained from the graph of log C versus t as shown in Fig 9.2.
Today, increased physiologic understanding of
pharmacokinetics shows that half-life is
a secondary parameter that depends
upon the primary parameters clearance and apparent volume of distribution according to following equation:
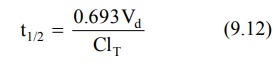
Apparent Volume of Distribution: The two separate
and independent pharmacokinetic characteristics
of a drug are –
1. Apparent volume of
distribution, and
2. Clearance.
Since these parameters are closely related with the physiologic
mechanisms in the body, they are called as primary parameters.
Modification of equation 9.7 defines apparent
volume of distribution:

Vd is a measure of the extent of
distribution of drug and is expressed in liters. The best and the simplest way
of estimating Vd of a drug is administering it by rapid i.v.
injection, determining the resulting plasma concentration immediately and using
the following equation:
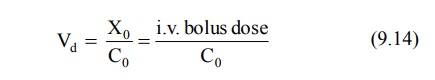
Equation 9.14 can only be used for drugs that obey
one-compartment kinetics. This is because the Vd can only be estimated
when distribution equilibrium is achieved between drug in plasma and that in
tissues and such equilibrium is established instantaneously for a drug that
follows one-compartment kinetics. A more general, more useful noncompartmental
method that can be applied to many compartment models for estimating the Vd
is:
For drugs given as i.v. bolus,
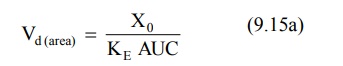
For drugs administered extravascularly (e.v.),
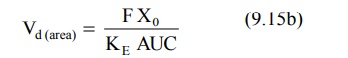
where, Xo = dose administered, and F =
fraction of drug absorbed into the systemic circulation. F is equal to one i.e. complete availability when the
drug is administered intravenously.
Clearance: Difficulties arise when one
applies elimination rate constant and half-life as pharmacokinetic parameters in an anatomical/physiological context
and as a measure of drug elimination mechanisms. A much more valuable
alternative approach for such applications is use of clearance parameters to
characterize drug disposition. Clearance
is the most important parameter in
clinical drug applications and is useful in evaluating the mechanism by which a
drug is eliminated by the whole organism or by a particular organ.
Just as Vd is needed to relate plasma
drug concentration with amount of drug in the body, clearance is a parameter to
relate plasma drug concentration with the rate of drug elimination according to
following equation:

Clearance is defined as the theoretical volume of body fluid containing drug (i.e. that fraction of
apparent volume of distribution) from
which the drug is completely removed in a given period of time. It is expressed in ml/min or liters/hour.
Clearance is usually further defined
as blood clearance (Clb),
plasma clearance (Clp) or
clearance based on unbound or free drug concentration (Clu)
depending upon the concentration C measured for the right side of the equation
9.17.
Total Body Clearance: Elimination
of a drug from the body involves processes occurring in kidney, liver, lungs and other eliminating organs. Clearance at an individual organ level is called as organ clearance. It can be estimated by dividing the rate of
elimination by each organ with the
concentration of drug presented to it. Thus,

The total body clearance, ClT, also called as total systemic clearance, is
an additive property of individual organ clearances. Hence,
Total Systemic Clearance
ClT = ClR + ClH + ClOthers
(9.18d)
Because of the additivity of clearance, the
relative contribution by any organ in eliminating a drug can be easily
calculated. Clearance by all organs other
than kidney is sometimes known as nonrenal clearance ClNR. It is the difference between total clearance and renal clearance.
According to an earlier definition (equation 9.17),
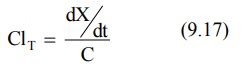
Substituting dX/dt = KEX from equation
9.3 in above equation, we get:
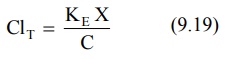
Since X/C = Vd (from equation 9.13), the equation 9.19 can be written as:
ClT = K E Vd (9.20a)
Parallel equations can be written for renal and
hepatic clearances as:
ClR = K e Vd (9.20b)
ClH = K m Vdm (9.20c)
Since KE = 0.693/t½ (from
equation 9.11), clearance can be related to half-life by the following
equation:
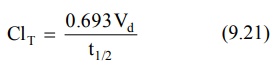
Identical equations can be written for ClR
and ClH in which cases the t½ will be urinary excretion
half-life for unchanged drug and metabolism half-life respectively. Equation
9.21. shows that as ClT decreases, as in renal insufficiency, t½
of the drug increases. As the ClT takes into account Vd,
changes in Vd as in obesity or oedematous condition will reflect
changes in ClT.
The noncompartmental method of computing total clearance for a drug that follows one-compartment kinetics is:

For a drug given by i.v. bolus, the renal clearance
ClR may be estimated by determining the total amount of unchanged
drug excreted in urine, Xu∞ and AUC.
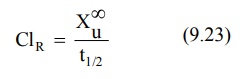
Organ Clearance: The best way of understanding
clearance is at individual organ level. Such
a physiologic approach is advantageous in predicting and evaluating the
influence of pathology, blood flow, P-D binding, enzyme activity, etc. on drug
elimination. At an organ level, the rate of elimination can be written as:

Substitution of equations 9.25 and 9.26 in equation
9.24 yields:
Rate of elimination = Q Cin
- Q Cout
(also called as Rate of extraction) = Q (Cin - Cout
) (9.27)
Division of above equation by concentration of drug
that enters the organ of elimination Cin yields an expression for
clearance of drug by the organ under consideration. Thus:
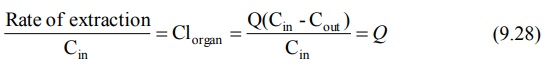
where, ER = (Cin – Cout)/Cin is called as extraction ratio. It has no units and its value ranges from zero (no elimination) to one (complete elimination). Based on ER values, drugs can be classified into 3 groups:
1. Drugs with high
ER (above 0.7),
2. Drugs with intermediate
ER (between 0.7 to 0.3), and
3. Drugs with low
ER (below 0.3).
ER is an index of how efficiently the eliminating organ clears the blood
flowing through it of drug. For example, an ER of 0.6 tells that 60% of the
blood flowing through the organ will be
completely cleared of drug. The fraction of drug that escapes removal by the organ is expressed as:
F=1-ER (9.29)
where, F
= systemic availability when the
eliminating organ is liver.
Hepatic Clearance: For certain drugs, the nonrenal
clearance can be assumed as equal to hepatic
clearance ClH. It is given as:
ClH =
ClT - ClR (9.30)
An equation parallel to equation 9.28 can also be
written for hepatic clearance:
ClH = QH ER H (9.31)
where,
QH = hepatic blood
flow (about 1.5 liters/min), and
ERH = hepatic
extraction ratio.
The hepatic clearance of drugs can be divided into
two groups:
1. Drugs with hepatic blood flow
rate-limited clearance, and
2. Drugs with intrinsic
capacity-limited clearance.
1. Hepatic Blood Flow: When ERH is one, ClH approaches its maximum value i.e. hepatic blood flow. In such a
situation, hepatic clearance is said to be perfusion
rate-limited or flow-dependent.
Alteration in hepatic blood flow significantly affects the elimination of drugs
with high ERH e.g. propranolol, lidocaine, etc. Such drugs are
removed from the blood as rapidly as they are presented to the liver (high
first-pass hepatic metabolism). Indocyanine green is so rapidly eliminated by
the human liver that its clearance is often used as an indicator of hepatic
blood flow rate. First-pass hepatic extraction is suspected when there is lack
of unchanged drug in systemic circulation after oral administration. Maximum oral availability F for such drugs can be computed from equation 9.29.
An extension of the same equation is
the noncompartmental method of estimating F:

TABLE 9.1
Influence of Blood Flow Rate and Protein Binding on Total Clearance of Drugs with High and with Low ER Values

On the contrary, hepatic blood flow has very little
or no effect on drugs with low ERH e.g. theophylline. For such
drugs, whatever concentration of drug present in the blood perfuses liver, is
more than what the liver can eliminate (low first-pass hepatic metabolism).
Similar discussion can be extended to the influence of blood flow on renal
clearance of drugs. This is illustrated in Table 9.1. Hepatic clearance of a
drug with high ER is independent of protein binding.
2. Intrinsic Capacity Clearance: Denoted as Clint, it
is defined as the inherent ability of an organ to irreversibly remove a drug in
the absence of any flow limitation. It depends, in this case, upon the hepatic enzyme activity. Drugs with low ERH
and with elimination primarily by
metabolism are greatly affected by changes in enzyme activity. Hepatic
clearance of such drugs is said to be capacity-limited,
e.g. theophylline. The t½ of such drugs show great intersubject variability. Hepatic clearance of
drugs with low ER is independent of blood flow rate but sensitive to changes in
protein binding.
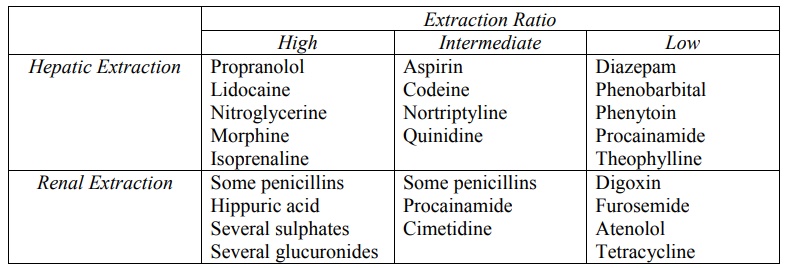
The hepatic and renal extraction ratios of some
drugs and metabolites are given in Table 9.2.
TABLE 9.2
Hepatic and Renal Extraction Ratio of Some Drugs and Metabolites
Related Topics