Reactions of the Cycle
| Home | | Biochemistry |Chapter: Biochemistry : Tricarboxylic Acid Cycle and Pyruvate Dehydrogenase Complex
In the TCA cycle, oxaloacetate is first condensed with an acetyl group from acetyl coenzyme A (CoA) and then is regenerated as the cycle is completed.
REACTIONS OF THE CYCLE
In the TCA cycle,
oxaloacetate is first condensed with an acetyl group from acetyl coenzyme A
(CoA) and then is regenerated as the cycle is completed (Figure 9.1).
Therefore, the entry of one acetyl CoA into one round of the TCA cycle does not
lead to the net production or consumption of intermediates. [Note: Two carbons
entering the cycle as acetyl CoA are balanced by two CO2 exiting.]
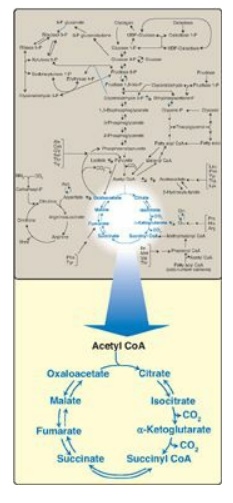
Figure 9.1 The tricarboxylic
acid cycle shown as a part of the essential pathways of energy metabolism. (See
Figure 8.2, for a more detailed view of the metabolic map.) CoA = coenzyme A.
A. Oxidative decarboxylation of pyruvate
The major source of acetyl CoA, the two-carbon substrate for the TCA cycle, is the oxidative decarboxylation of pyruvate. Pyruvate, the end product of aerobic glycolysis, must be transported from the cytosol into the mitochondrion. This is accomplished by a specific transporter that facilitates movement of pyruvate across the inner mitochondrial membrane. Once in the mitochondrial matrix, pyruvate is converted to acetyl CoA by the pyruvate dehydrogenase complex (PDH complex), which is a multienzyme complex. [Note: Strictly speaking, the PDH complex is not part of the TCA cycle, but it supplies substrate for the cycle.]
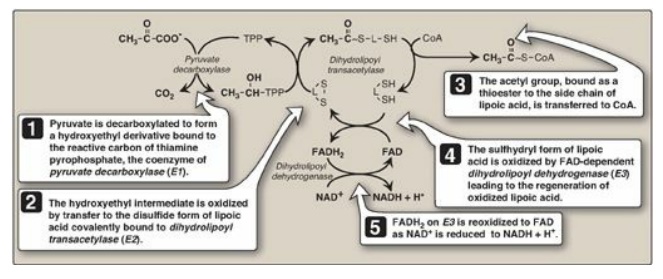
Figure 9.2 Mechanism of action of the pyruvate dehydrogenase complex. [Note: All the coenzymes of the complex, except for lipoic acid, are derived from vitamins. TPP is from thiamine, FAD from riboflavin, NAD from niacin, and CoA from pantothenic acid.] TPP = thiamine pyrophosphate; L = lipoic acid; CoA = coenzyme A; FAD(H2) = flavin adenine dinucleotide; NAD(H) = nicotinamide adenine dinucleotide.
1. Component enzymes: The PDH complex is a protein
aggregate of multiple copies of three enzymes, pyruvate carboxylase (E1,
sometimes called pyruvate dehydrogenase), dihydrolipoyl transacetylase (E2),
and dihydrolipoyl dehydrogenase (E3). Each catalyzes a part of the overall
reaction (Figure 9.2). Their physical association links the reactions in proper
sequence without the release of intermediates. In addition to the enzymes
participating in the conversion of pyruvate to acetyl CoA, the complex also
contains two tightly bound regulatory enzymes, pyruvate dehydrogenase kinase
(PDH kinase) and pyruvate dehydrogenase phosphatase (PDH phosphatase).
2. Coenzymes: The PDH complex contains five coenzymes that act as
carriers or oxidants for the intermediates of the reactions shown in Figure
9.2. E1 requires thiamine pyrophosphate (TPP), E2 requires lipoic acid and CoA,
and E3 requires flavin adenine dinucleotide (FAD) and nicotinamide adenine
dinucleotide (NAD+). [Note: TPP, lipoic acid, and FAD are tightly
bound to the enzymes and function as coenzymes-prosthetic groups.]
Deficiencies of thiamine or niacin can cause serious central nervous system problems. This is because brain cells are unable to produce sufficient ATP (via the TCA cycle) if the PDH complex is inactive. Wernicke-Korsakoff, an encephalopathy-psychosis syndrome due to thiamine deficiency, may be seen with alcohol abuse.
3. Regulation of the pyruvate dehydrogenase
complex:
Covalent modifications by the two regulatory enzymes that are part of the
complex alternately activate and inactivate E1. The cyclic AMP–independent PDH
kinase phosphorylates and, thereby, inactivates E1, whereas PDH phosphatase dephosphorylates
and activates E1 (Figure 9.3). The kinase itself is allosterically activated by
ATP, acetyl CoA, and NADH. Therefore, in the presence of these high-energy
signals, the PDH complex is turned off. [Note: It is actually the rise in the
ATP/ADP, NADH/NAD +, or acetyl CoA/CoA ratios that affects enzymic activity.]
Pyruvate is a potent inhibitor of PDH kinase. Thus, if pyruvate concentrations
are elevated, E1 will be maximally active. Calcium (Ca2+) is a
strong activator of PDH phosphatase, stimulating E1 activity. This is
particularly important in skeletal muscle, where release of Ca2+
during contraction stimulates the PDH complex and, thereby, energy production.
[Note: Although covalent regulation by the kinase and phosphatase is primary,
the complex is also subject to product (NADH and acetyl CoA) inhibition.]
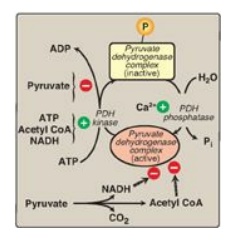
Figure 9.3 Regulation of
pyruvate dehydrogenase (PDH) complex. [ → denotes product inhibition.]
4. Pyruvate dehydrogenase complex deficiency: A deficiency in the activity of
the α subunit of the dimeric E1 component of the PDH
complex, although rare, is the most common biochemical cause of congenital
lactic acidosis. This enzyme deficiency results in an inability to convert
pyruvate to acetyl CoA, causing pyruvate to be shunted to lactate via lactate
dehydrogenase. This creates particular problems for the brain, which relies on
the TCA cycle for most of its energy and is particularly sensitive to acidosis.
Symptoms are variable and include neurodegeneration; muscle spasticity; and, in
the neonatal onset form, early death. The gene for the α subunit is X linked,
and, because both males and females may be affected, the deficiency is
classified as X-linked dominant. Although there is no proven treatment for PDH
complex deficiency, dietary restriction of carbohydrate and supplementation
with thiamine may reduce symptoms in select patients.
Leigh syndrome (subacute necrotizing
encephalomyelopathy) is a rare, progressive, neurodegenerative disorder caused
by defects in mitochondrial ATP production, primarily as a result of mutations
in genes that code for proteins of the PDH complex, the electron transport
chain, or ATP synthase. Both nuclear and mitochondrial DNA can be affected.
5. Mechanism of arsenic poisoning: As previously described, pentavalent arsenic (arsenate) can interfere with glycolysis at the glyceraldehyde 3-phosphate step, thereby decreasing ATP production. “Arsenic poisoning” is, however, due primarily to inhibition of enzymes that require lipoic acid as a coenzyme, including E2 of the PDH complex, α-ketoglutarate dehydrogenase (see below), and branched-chain α-keto acid dehydrogenase. Arsenite (the trivalent form of arsenic) forms a stable complex with the thiol (–SH) groups of lipoic acid, making that compound unavailable to serve as a coenzyme. When it binds to lipoic acid in the PDH complex, pyruvate (and, consequently, lactate) accumulates. As with PDH complex deficiency, this particularly affects the brain, causing neurologic disturbances and death.
B. Synthesis of citrate from acetyl coenzyme A and oxaloacetate
The condensation of
acetyl CoA and oxaloacetate (OAA) to form citrate (a tricarboxylic acid) is
catalyzed by citrate synthase (Figure 9.4). This aldol condensation has an
equilibrium far in the direction of citrate synthesis. In humans, citrate
synthase is not an allosteric enzyme. It is inhibited by its product, citrate.
Substrate availability is another means of regulation for citrate synthase. The
binding of OAA causes a conformational change in the enzyme that generates a
binding site for acetyl CoA. [Note: Citrate, in addition to being an
intermediate in the TCA cycle, provides a source of acetyl CoA for the
cytosolic synthesis of fatty acids. Citrate also inhibits phosphofructokinase-1
(PFK-1), the rate-limiting enzyme of glycolysis, and activates acetyl CoA
carboxylase (the rate-limiting enzyme of fatty acid synthesis;).]
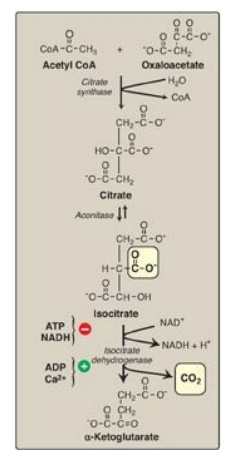
Figure 9.4 Formation of α-ketoglutarate from acetyl coenzyme A (CoA) and oxaloacetate. NAD(H) = nicotinamide adenine dinucleotide
C. Isomerization of citrate
Citrate is isomerized
to isocitrate by aconitase (aconitate hydratase), an Fe-S protein (see Figure
9.4). [Note: Aconitase is inhibited by fluoroacetate, a plant toxin that is
used as a pesticide. Fluoroacetate is converted to fluoroacetyl CoA, which
condenses with OAA to form fluorocitrate (a potent inhibitor of aconitase),
resulting in citrate accumulation.]
D. Oxidative decarboxylation of isocitrate
Isocitrate
dehydrogenase catalyzes the irreversible oxidative decarboxylation of
isocitrate, yielding the first of three NADH molecules produced by the cycle
and the first release of CO2 (see Figure 9.4). This is one of the
rate-limiting steps of the TCA cycle. The enzyme is allosterically activated by
ADP (a low-energy signal) and Ca2+ and is inhibited by ATP and NADH,
levels of which are elevated when the cell has abundant energy stores.
E. Oxidative decarboxylation of α-ketoglutarate
The conversion of
α-ketoglutarate to succinyl CoA is catalyzed by the α-ketoglutarate dehydrogenase
complex, a protein aggregate of multiple copies of three enzymes (Figure 9.5).
The mechanism of this oxidative decarboxylation is very similar to that used
for the conversion of pyruvate to acetyl CoA by the PDH complex. The reaction
releases the second CO2 and produces the second NADH of the cycle.
The coenzymes required are TPP, lipoic acid, FAD, NAD+, and CoA.
Each functions as part of the catalytic mechanism in a way analogous to that
described for the PDH complex. The equilibrium of the reaction is far in the
direction of succinyl CoA, a high-energy thioester similar to acetyl CoA.
α-Ketoglutarate dehydrogenase complex is inhibited by its products, NADH and
succinyl CoA, and activated by Ca2+. However, it is not regulated by
phosphorylation/dephosphorylation reactions as described for PDH complex.
[Note: α-Ketoglutarate is also produced by the oxidative deamination and
transamination of the amino acid glutamate.]
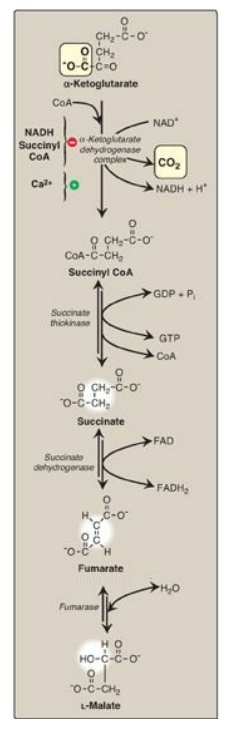
Figure 9.5 Formation of
malate from α-ketoglutarate. NAD(H) = nicotinamide adenine dinucleotide; GDP =
guanosine diphosphate; P = phosphate; CoA = coenzyme A; FAD(H2) =
flavin adenine dinucleotide.
F. Cleavage of succinyl coenzyme A
Succinate thiokinase
(also called succinyl CoA synthetase, named for the reverse reaction) cleaves
the high-energy thioester bond of succinyl CoA (see Figure 9.5). This reaction
is coupled to phosphorylation of guanosine diphosphate (GDP) to guanosine
triphosphate (GTP). GTP and ATP are energetically interconvertible by the
nucleoside diphosphate kinase reaction:
GTP + ADP ↔ GDP +
ATP
The generation of GTP by succinate thiokinase is another example of substrate-level phosphorylation. [Note: Succinyl CoA is also produced from propionyl CoA derived from the metabolism of fatty acids with an odd number of carbon atoms, and from the metabolism of several amino acids.]
G. Oxidation of succinate
Succinate is oxidized
to fumarate by succinate dehydrogenase, as FAD (its coenzyme) is reduced to FADH2
(see Figure 9.5). Succinate dehydrogenase is the only enzyme of the TCA cycle
that is embedded in the inner mitochondrial membrane. As such, it functions as
Complex II of the electron transport chain. [Note: FAD, rather than NAD+,
is the electron acceptor because the reducing power of succinate is not
sufficient to reduce NAD+.]
H. Hydration of fumarate
Fumarate is hydrated to
malate in a freely reversible reaction catalyzed by fumarase (fumarate
hydratase; see Figure 9.5). [Note: Fumarate is also produced by the urea cycle,
in purine synthesis, and during catabolism of the amino acids phenylalanine and
tyrosine.]
I. Oxidation of malate
Malate is oxidized to
oxaloacetate by malate dehydrogenase (Figure 9.6). This reaction produces the
third and final NADH of the cycle. The standard free energy change (∆G0;)
of the reaction is positive, but the reaction is driven in the direction of OAA
by the highly exergonic citrate synthase reaction. [Note: OAA is also produced
by the transamination of the amino acid aspartic acid.]
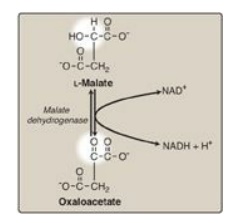
Figure 9.6 Formation (regeneration) of oxaloacetate from malate. NAD(H) = nicotinamide adenine dinucleotide.
Related Topics
