Ring Construction
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Planning Organic Syntheses
The use of carbonyl groups to set the polarity of bond disconnections in retrosynthetic analysis is useful for the construction of rings as well.
RING CONSTRUCTION
The
use of carbonyl groups to set the polarity of bond disconnections in
retrosynthetic analysis is useful for the construction of rings as well. If a
carbon electrophile and a carbon nucleophile are connected by a carbon chain,
they can react with each other to form a carbon–carbon bond. This is an
absolutely nor-mal type of carbon–carbon bond-forming process, but the fact
that the carbon nucleophile and carbon electrophile are connected by a chain means
that the new carbon–carbon bond closes up the ends of the chain, forming a
ring.
For
example, the Claisen reaction is a reaction of an ester enolate with an ester
to produce a β ketoester. We learned
this reaction earlier.
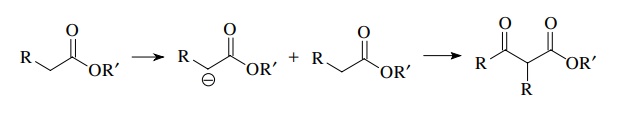
If
both ester groups are in the same molecule and are connected by a chain, then a
Claisen-type reaction between the α
position of one ester and the carbonyl group of the other gives a new
carbon–carbon bond and closes up the ring. (This reaction is actually called
the Dieckmann condensation, but it is nothing more than an intramolecular
Claisen reaction.)
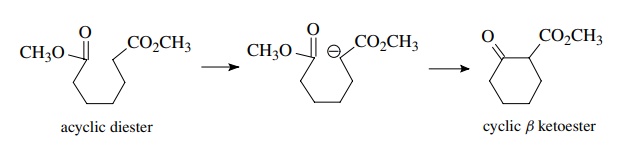
Ring-forming
reactions are very important in retrosynthetic analysis because many
interesting targets are cyclic compounds and often rings must be installed
rather than being present in the starting materials. From a retrosynthetic
point of view, there is really no difference between ring-forming reactions and
other carbon–carbon bond-forming reactions. One looks for the same polarities
and functional group features as in acyclic systems.
The
only thing that is different is that some rings are more easily formed than
others. The rule of thumb is that rings of three, five, and six members are
routinely formed, while rings of four or more than six members are formed with
greater difficulty. For example, reaction of diethyl malonate with
1,2-dibromoethane and two equivalents of base gives diethyl cyclopropane-1,1-dicarboxylate
in high yield. Ring formation occurs by a double-displacement sequence.
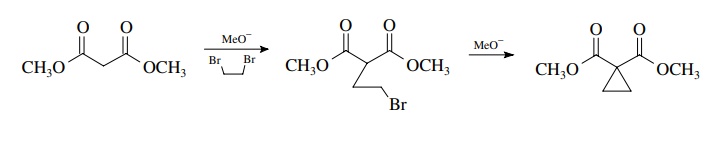
Likewise
reactions with 1,3-dibromopropane, 1,4-dibromobutane, or 1,5-dibromopentane
give the corresponding cyclobutyl-, cyclopentyl-, and cyclo-hexyl-1,1-dicarboxylates.
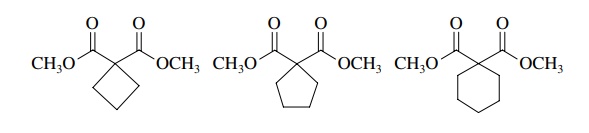
The
yields of these reactions are not the same, however, and reactions which
produce three-, five-, and six-membered rings are generally more effective. Use
of 1,6-dibromohexane fails to give the cycloheptyl product.
Ring
closure requires that a reactive center at one end of the chain encoun-ters a
reactive center at the other end of the
same chain in the bond-forming process. The alternative is for the reactive
center on one chain to react with a reactive center of a different chain. The
first case produces a ring, the second case a polymer. An analogy is a chain
with complementary hooks at each end representing electrophilic and
nucleophilic carbons at the ends of the chain. If two hooks on the same chain
link up, a ring is formed, whereas if hooks from different chains link up, a
larger molecule is formed with hooks remaining on each end. These can link up
further to form progressively larger molecules.
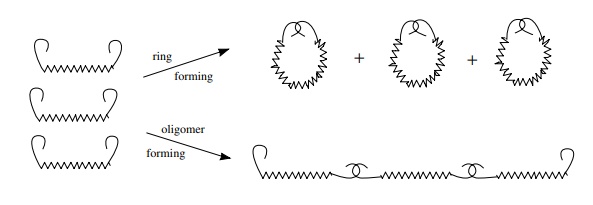
For
short chains which would give three- to six-membered rings upon ring closure,
there is a higher probability that one end of the chain will encounter the
other end of the same chain and react intramolecularly
before it will encounter the end of another chain and react intermolecularly. Thus ring closure is
normally favored over oligomerization for smaller rings of three to six
members. On the other hand, as the chains become longer, it becomes less likely
that the end of a chain will encounter the other end of the same chain before
it encounters and reacts with the end of another chain. The break point is
between six-membered rings, which are formed readily, and seven-membered rings,
which are not easily formed. While this reasoning is a great simplification, it
suffices to provide a good working model to predict the success for
ring-forming reactions.
We
can use this model in retrosynthetic analysis quite successfully. Suppose one
were asked to produce cyclohexanone C
from acyclic starting materials.
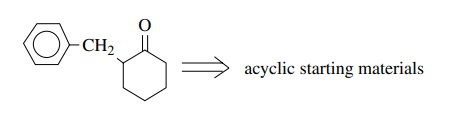
In this monofunctional compound, the ketone could serve as an electrophilic center in a cyclization step. Disconnection at the indicated bond leads to the polarity shown; however, it is immediately obvious that the carbon nucleophile occurs at an unactivated position, and there is no good way to produce it there without a control element at that position.
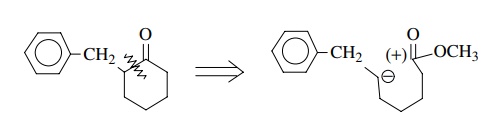
However, use of an ester group could activate this position toward anion formation and thus we could write instead
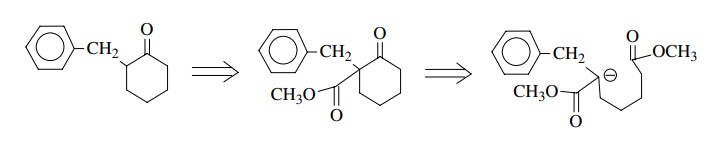
Now
all is well in terms of polarity and we recognize this as a Dieckmann reaction
followed by hydrolysis and decarboxylation of the β-ketoester product. Proceeding backward we write
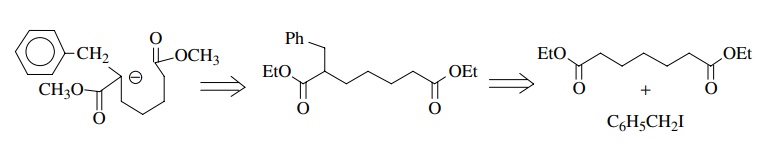
Alkylation
of diethyl suberate with benzyl iodide would produce the α-benzylated product which would cyclize in the presence of base.
This particular target does not present a regiochemical difficulty. Base could
pull off either α proton and two
different enolates would be produced; however, both enolates cyclize to give
the same product after decarboxylation.
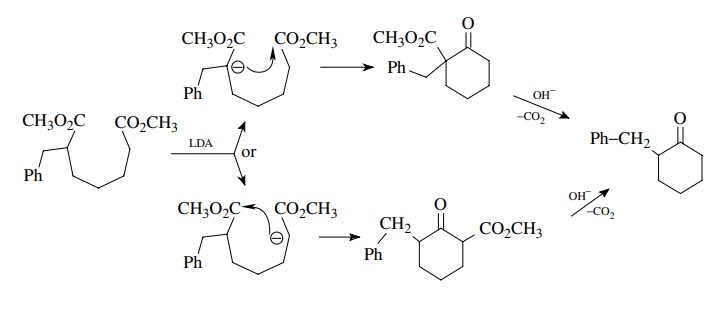
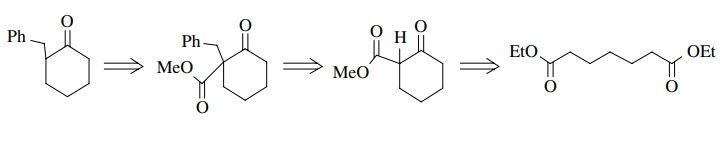
If
molecular symmetry were included in the cyclization precursor, then we would
not have to worry about regiochemistry in the ring closure. Noting that the
product of ring formation is a β
ketoester, which itself is a good carbon nucleophile, an alternate
retrosynthesis (which actually is much better) is the following in which the
benzyl group is added after the ring is formed (in the forward synthesis):
Related Topics
