Carbon Skeleton Synthesis
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Planning Organic Syntheses
In general, it is most efficient to construct the carbon skeleton first and then adjust the functionality to give the target.
CARBON SKELETON SYNTHESIS
In
general, it is most efficient to construct the carbon skeleton first and then
adjust the functionality to give the target. Thus in retrosynthetic analysis we
most often move backward from the target to compounds which contain functional
groups important in carbon–carbon bond-making reactions. As a consequence
carbonyl groups play a very important role in retrosynthetic analysis. They are
very useful sources of both electrophilic and nucleophilic carbon that can be
used in making carbon–carbon bonds. Based on our earlier discussions, a
carbonyl group can be seen to influence the polarity of nearby carbons as
shown.
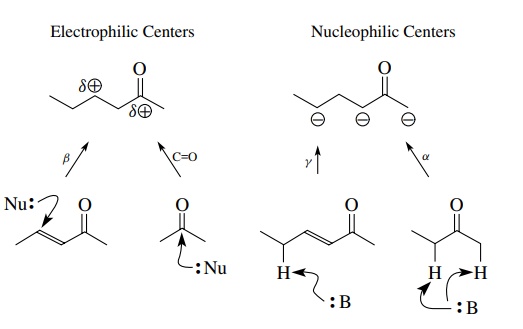
By
bond polarity and resonance, the carbonyl carbon and a carbon β to the carbonyl carbon can be utilized
as electrophilic centers—the carbonyl group by direct nucleophilic addition and
the β carbon by Michael addition to
an α,β-unsaturated ketone. By
resonance interaction, the α position
in carbonyl compounds and γ positions
in α,β-unsaturated carbonyl compounds can be con-verted to nucleophilic
centers by proton removal. These “normal” polarities are used frequently in
retrosynthetic planning as points of disconnection to establish potential
bond-forming steps using carbonyl groups.
As
an initial exercise consider the synthesis of C from cyclohexyl bromide. First one has to note relevant facts
about the target, such as (a) it contains an ester and (b) it has two more
carbons than the starting material so a two-carbon fragment will have to be
attached by a new carbon–carbon bond.

Looking
at the starting material, it is noted that it contains an electrophilic carbon;
thus the needed carbon–carbon bond could be formed by reaction of a two-carbon nucleophile
with the electrophilic center of bromocyclohexane. Noting that the ester group
needed in the product acidifies the α
position, it could be used to make the nucleophilic carbon required for
carbon–carbon bond formation. Retrosynthetically this can be written as
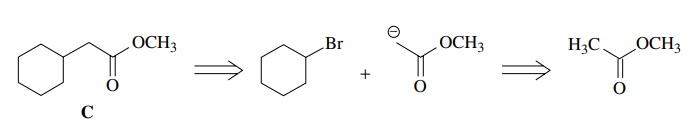
So
the synthesis could be done in one step by making the anion of methyl acetate
and reacting it with bromocyclohexane. The polarities of the reaction partners
match nicely, but the problem is that alkylations of secondary bromides with
enolates often give poor yields. The enolate is a strong base, which pro-motes
elimination in the secondary bromide rather than giving the substitution
product needed in the synthesis. Thus elimination from cyclohexyl bromide to cyclohexene
would be a major process if the reaction were attempted. While the
retrosynthetic step seems reasonable, the synthetic step has known
difficulties. It is important to work backward in the retrosynthetic analysis
and then check each forward step for validity.
What
is needed in the synthesis of C is a
two-carbon nucleophile (or its equivalent) which is less basic than an enolate
so elimination is not competitive. If product C is recognized as an acetic acid derivative, then the following
analysis can be made. A malonate ion used as the carbon nucleophile is much
less basic than a simple ester enolate and hence undergoes substitution readily
but does not promote elimination effectively, particularly in secondary
systems.
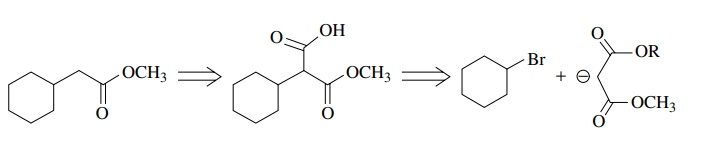
Now
clearly there will have to be some functional group adjustment in the synthesis
because a hydrolysis of the alkylated malonate must be carried out in order to
give decarboxylation to the acetic ester derivative. Either an unsymmetric
malonate must be used that can be differentially hydrolyzed or both ester
functions of the malonate could be hydrolyzed and after decarboxylation the
acid could be reesterified. The actual synthesis could be planned as shown in
(10.1) or (10.2):
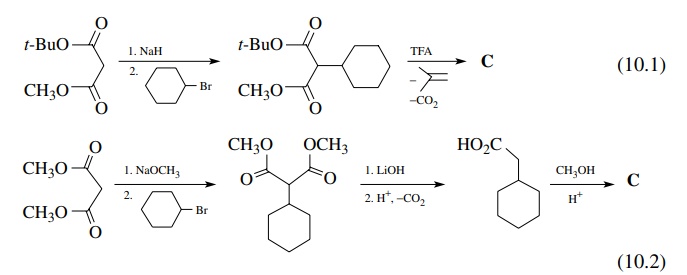
Synthesis
(10.2) contains an extra step but uses very cheap and available starting
materials. Synthesis (10.1) is shorter and goes in high yield but requires
anhydrous conditions for the alkylation and a more expensive malonate starting
material.
The
use of Pd(0) to form carbon–carbon bonds was shown to be very effective in many
cases. Could one of the coupling methods catalyzed by Pd(0) be used to attach
the two-carbon fragment needed to construct the skeleton of C? Since we are restricted to
cyclohexyl bromide as the starting material, direct reaction with Pd(0) is not
feasible because Pd(0) does not give oxidative addition with saturated
bromides. Moreover saturated bromides do not undergo transmetallation with
Pd(0), so it could not serve as the second component in a Pd(0)-catalyzed
coupling. Thus the reactivity requirements of Pd(0)-catalyzed coupling
reactions are incompatible with the starting material and thus are not usable
for the present construction.
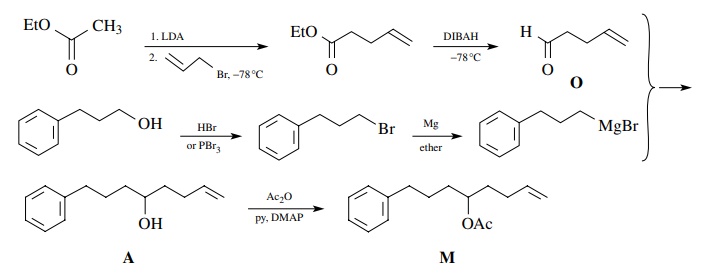
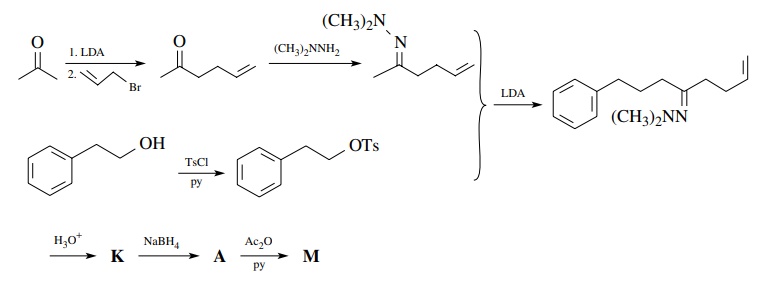
Next
consider the synthesis of M from
“readily available” starting materials. The relevant facts about M are that (a) it contains an aromatic
ring, an acetate ester, and a vinyl group; (b) it has a straight chain attached
to the ring; and (c) all the functional groups are isolated.
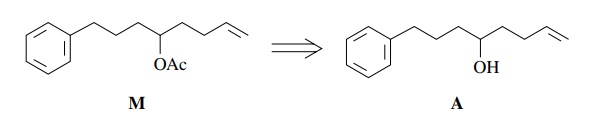
To
begin the retrosynthetic analysis, note that the acetate ester is easily
pro-duced from the corresponding alcohol A.
Therefore conversion of A to M using acetic anhydride/pyridine could
be used in the synthetic step. (Remember: For each retrosynthetic step, a
reaction must be available to accomplish the syn-thetic step.)
Now
the alcohol functional group in A is
a natural point for bond discon-nection to take place since alcohols are the
products of carbon nucleophiles and carbonyl groups. If we consider bond a in our retrosynthetic analysis, then
the next retrosynthetic step would be
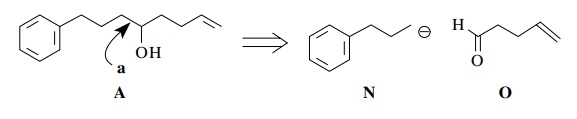
Since
the anion N is a nonstabilized
carbanion, an organometallic nucleophile such as an organolithium or a Grignard
reagent could be prepared from the corresponding bromide.
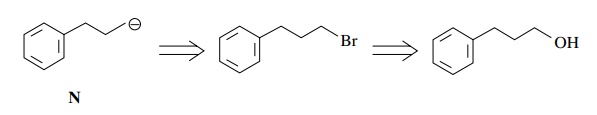
The
bromide could be prepared from 3-phenyl-1-propanol ($59/kg). The unsaturated
aldehyde O can be made by oxidation
(CrO3.py) of 4-penten-1-ol ($41.80/10 g). A cheaper way is to make
ethyl 4-pentenoate from ethyl acetate ($15/gal) and allyl bromide ($19/100 g)
and reduce it to the aldehyde O with
DIBAH ($19/ 0.1 mol).
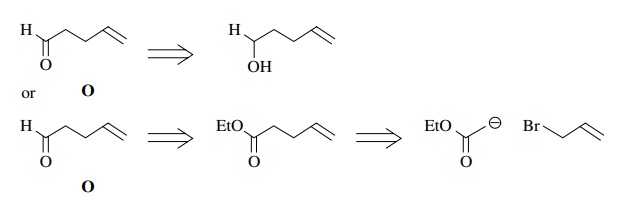
Thus
a synthesis of M based on this
retrosynthetic analysis would start with ethyl acetate, allyl bromide, and
3-phenyl-1-propanol.
Now
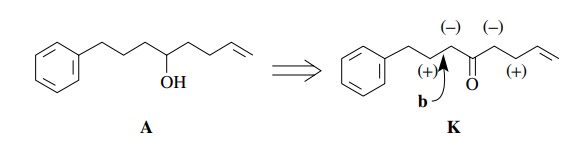
we go back to A and consider
disconnection at a different bond. Suppose we recognize that alcohol A could easily come from reduction of
ketone K.
Now
considering the polarities possible, a great number of disconnections can be
envisioned. Choosing bond b means
that polarity (with respect to the carbonyl group) would be
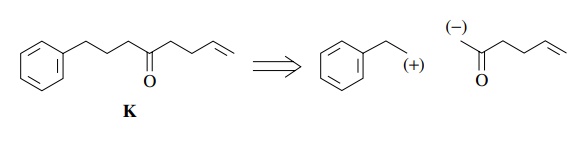
and
thus an enolate reacting with a carbon electrophile would be appropriate. A
valid retrosynthetic step would be
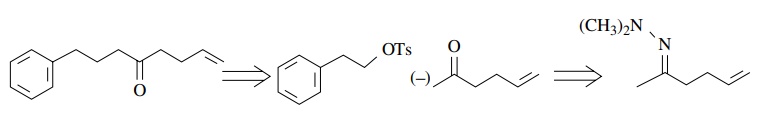
Because
the tosylate is primary, substitution should be the major pathway (although in
this case elimination could be problematic because of conjugation with the
phenyl ring). We note, however, that the enolate needed is the kinetic enolate
of 5-hexen-2-one. This poses a regiochemical control problem which can be
solved by making the N ,N -dimethylhydrazone of the ketone. The
ketone 5-hexene-2-one is available ($46.20/25 g) or can be made by allylation
of acetone.
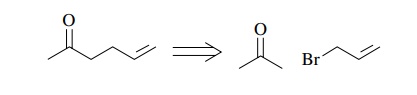
Thus
a synthesis based on this retrosynthetic analysis starts with β-phenylethanol ($35.60/kg), acetone,
and allyl bromide. This route is comparable to the first in both number of
steps and cost. It differs in that regiochemical control of enolate formation
is a crucial feature. Several other syntheses of K can be devised by other disconnections suggested by the natural
polarities engendered by the ketone group.
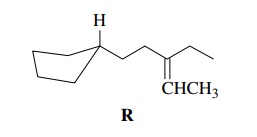
Next
consider compound R. When the
relevant facts are considered, we see that R
is merely an olefin with a saturated ring present. Because of the five-membered
ring and because the target has 12 carbon atoms, it is unlikely that compounds
with the carbon skeleton of R will
be available commercially; hence carbon–carbon bond-forming reactions will be
needed to assemble the carbon skeleton.
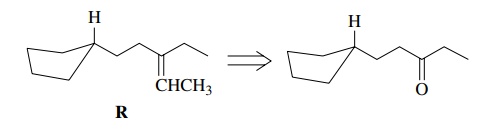
Moreover
the carbon–carbon double bond is a natural starting point for bond
disconnection. A logical retrosynthetic step would be disconnection to a ketone
because a Wittig reaction could be used to convert the ketone to the ethylidene
product. (Note that dehydration of an alcohol to the olefin is not a viable
synthetic step because dehydration would lead to a mixture of trisubstituted
olefins.)
Once
the ketone is recognized as a useful intermediate, normal polarities can be
used to disconnect it retrosynthetically. For example, a good disconnection
could be as shown, where Michael addition to ethyl vinyl ketone by a
cyclopentyl anion would give the needed ketone.
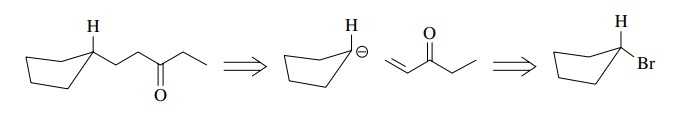
The
cyclopentyl nucleophile, which should be an organocuprate to ensure Michael
addition, could be produced from cyclopentyl bromide. The synthetic sequence
consistent with the retrosynthetic analysis turns out to be a rather simple
synthesis of what at first sight is a more difficult molecule.
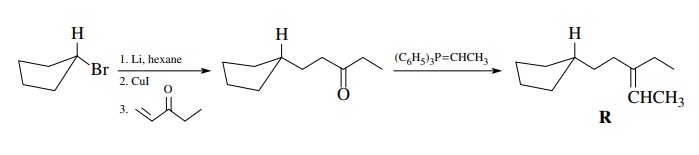
Now
there are a variety of other ways to disconnect R in the retrosynthetic analysis. As long as each synthetic step is
valid and the target can be produced by the proposed synthetic route, then it
is a correct solution. There can be many correct synthetic solutions for a
given target and the “best” one may depend on factors other than those related
strictly to the synthetic viability. Availability of starting materials,
disposal of reaction by-products, number of steps, reagent sen-sitivity,
expected yields, number of purifications, and the stereochemistry (among
others) all contribute to the evaluation of a synthetic route.
Related Topics
