Steps in Prokaryotic DNA Synthesis
| Home | | Biochemistry |Chapter: Biochemistry : DNA Structure, Replication, and Repair
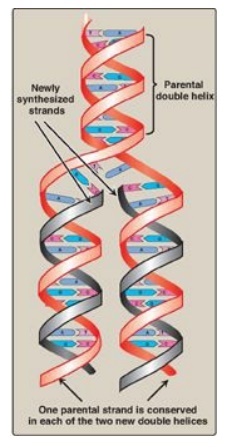
When the two strands of the DNA double helix are separated, each can serve as a template for the replication of a new complementary strand.
STEPS IN PROKARYOTIC DNA SYNTHESIS
When the two strands of
the DNA double helix are separated, each can serve as a template for the
replication of a new complementary strand. This produces two daughter
molecules, each of which contains two DNA strands with an antiparallel
orientation (see Figure 29.3). This process is called semiconservative
replication because, although the parental duplex is separated into two halves
(and, therefore, is not “conserved” as an entity), each of the individual
parental strands remains intact in one of the two new duplexes (Figure 29.8).
The enzymes involved in the DNA replication process are template-directed
polymerases that can synthesize the complementary sequence of each strand with
extraordinary fidelity. The reactions described in this section were first
known from studies of the bacterium Escherichia coli (E. coli), and the
description given below refers to the process in prokaryotes. DNA synthesis in
higher organisms is more complex but involves the same types of mechanisms. In
either case, initiation of DNA replication commits the cell to continue the
process until the entire genome has been replicated.
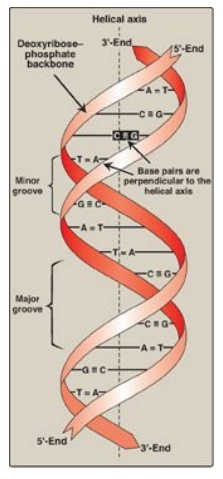
Figure 29.3 DNA double helix, illustrating some of its major structural features.
A. Separation of the two complementary DNA strands
In order for the two
strands of the parental dsDNA to be replicated, they must first separate (or
“melt”) over a small region, because the polymerases use only ssDNA as a
template. In prokaryotic organisms, DNA replication begins at a single, unique
nucleotide sequence, a site called the origin of replication, or ori (Figure
29.9A). [Note: This sequence is referred to as a consensus sequence, because
the order of nucleotides is essentially the same at each site.] The ori
includes short, AT-rich segments that facilitate melting. In eukaryotes,
replication begins at multiple sites along the DNA helix (Figure 29.9B). Having
multiple origins of replication provides a mechanism for rapidly replicating
the great length of eukaryotic DNA molecules.
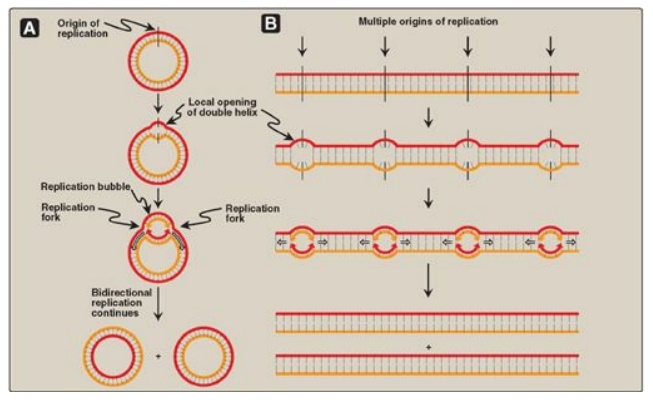
Figure 29.9 Replication of DNA: origins and replication forks. A. Small prokaryotic circular DNA. B. Very long eukaryotic DNA.
B. Formation of the replication fork
As the two strands
unwind and separate, synthesis occurs at two replication forks that move away
from the origin in opposite directions (bidirectionally), generating a replication
bubble (see Figure 29.9). [Note: The term “replication fork” derives from the
Y-shaped structure in which the tines of the fork represent the separated
strands (Figure 29.10).]
1. Proteins required for DNA strand separation: Initiation of DNA replication
requires the recognition of the origin by a group of proteins that form the
prepriming complex. These proteins are responsible for maintaining the
separation of the parental strands, and for unwinding the double helix ahead of
the advancing replication fork. These proteins include the following.
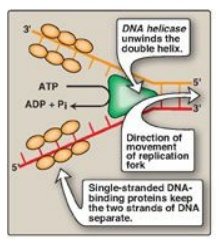
Figure 29.10 Proteins responsible for maintaining the separation of the parental strands and unwinding the double helix ahead of the advancing replication fork ![]() . ADP = adenosine diphosphate; Pi = inorganic phosphate.
. ADP = adenosine diphosphate; Pi = inorganic phosphate.
a. DnaA protein: DnaA protein binds to specific nucleotide
sequences (DnaA boxes) within the origin of replication, causing the short,
tandemly arranged (one after the other) AT-rich regions in the origin to melt.
Melting is adenosine triphosphate (ATP) dependent, and results in strand
separation with the formation of localized regions of ssDNA.
b. DNA helicases: These enzymes bind to ssDNA near
the replication fork and then move into the neighboring double-stranded region,
forcing the strands apart (in effect, unwinding the double helix). Helicases
require energy provided by ATP (see Figure 29.10). Unwinding at the replication
fork causes supercoiling in other regions of the DNA molecule. [Note: DnaB is
the principal helicase of replication in E. coli. Its binding to DNA requires
DnaC.]
c. Single-stranded DNA-binding protein: This protein binds to the ssDNA
generated by helicases (see Figure 29.10). Binding is cooperative (that is, the
binding of one molecule of single-stranded binding [SSB] protein makes it
easier for additional molecules of SSB protein to bind tightly to the DNA
strand). The SSB proteins are not enzymes, but rather serve to shift the
equilibrium between dsDNA and ssDNA in the direction of the single-stranded
forms. These proteins not only keep the two strands of DNA separated in the
area of the replication origin, thus providing the single-stranded template
required by polymerases, but also protect the DNA from nucleases that degrade
ssDNA.
2. Solving the problem of supercoils: As the two strands of the double
helix are separated, a problem is encountered, namely, the appearance of
positive supercoils in the region of DNA ahead of the replication fork as a
result of overwinding (Figure 29.11), and negative supercoils in the region
behind the fork. The accumulating positive supercoils interfere with further
unwinding of the double helix. [Note: Supercoiling can be demonstrated by
tightly grasping one end of a helical telephone cord while twisting the other
end. If the cord is twisted in the direction of tightening the coils, the cord
will wrap around itself in space to form positive supercoils. If the cord is
twisted in the direction of loosening the coils, the cord will wrap around
itself in the opposite direction to form negative supercoils.] To solve this
problem, there is a group of enzymes called DNA topoisomerases, which are
responsible for removing supercoils in the helix by transiently cleaving one or
both of the DNA strands.
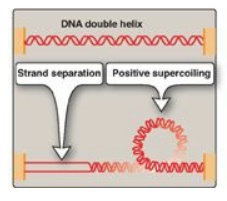
Figure 29.11 Positive
supercoiling resulting from DNA strand separation.
a. Type I DNA topoisomerases: These enzymes reversibly cleave
one strand of the double helix. They have both strand-cutting and
strand-resealing activities. They do not require ATP, but rather appear to
store the energy from the phosphodiester bond they cleave, reusing the energy
to reseal the strand (Figure 29.12). Each time a transient “nick” is created in
one DNA strand, the intact DNA strand is passed through the break before it is
resealed, thus relieving (“relaxing”) accumulated supercoils. Type I
topoisomerases relax negative supercoils (that is, those that contain fewer
turns of the helix than relaxed DNA) in E. coli, and both negative and positive
supercoils (that is, those that contain fewer or more turns of the helix than
relaxed DNA) in many prokaryotic cells (but not E. coli) and in eukaryotic
cells.
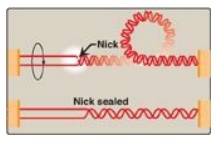
Figure 29.12 Action of type I
DNA topoisomerases.
b. Type II DNA topoisomerases: These enzymes bind tightly to the
DNA double helix and make transient breaks in both strands. The enzyme then
causes a second stretch of the DNA double helix to pass through the break and,
finally, reseals the break (Figure 29.13). As a result, both negative and
positive supercoils can be relieved by this ATP-requiring process. DNA gyrase,
a type II topoisomerase found in bacteria and plants, has the unusual property
of being able to introduce negative supercoils into circular DNA using energy from
the hydrolysis of ATP. This facilitates the replication of DNA because the
negative supercoils neutralize the positive supercoils introduced during
opening of the double helix. It also aids in the transient strand separation
required during transcription.
Anticancer agents, such as the camptothecins,
target human type I topoisomerases, whereas etoposide targets human type II
topoisomerases. Bacterial DNA gyrase is a unique target of a group of
antimicrobial agents called fluoroquinolones (for example, ciprofloxacin).
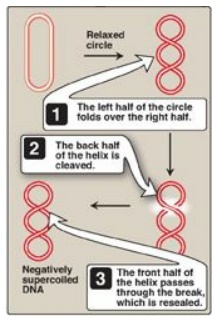
Figure 29.13 Action of type II
DNA topoisomerase.
C. Direction of DNA replication
The DNA polymerases
responsible for copying the DNA templates are only able to “read” the parental
nucleotide sequences in the 3I →5 I direction, and they synthesize the
new DNA strands only in the 5 I →3 I (antiparallel) direction. Therefore,
beginning with one parental double helix, the two newly synthesized stretches
of nucleotide chains must grow in opposite directions, one in the 5 I →3 I direction toward the replication fork and one in the 5 I →3 I direction away from the replication fork (Figure 29.14). This
feat is accomplished by a slightly different mechanism on each strand.
1. Leading strand: The strand that is being copied in
the direction of the advancing replication fork is called the leading strand
and is synthesized continuously.
2. Lagging strand: The strand that is being copied in
the direction away from the replication fork is synthesized discontinuously,
with small fragments of DNA being copied near the replication fork. These short
stretches of discontinuous DNA, termed Okazaki fragments, are eventually joined
(ligated) to become a single, continuous strand. The new strand of DNA produced
by this mechanism is termed the lagging strand.
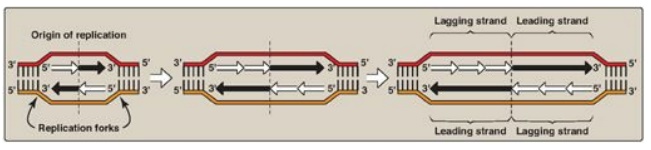
Figure 29.14 Discontinuous synthesis of DNA.
D. RNA primer
DNA polymerases cannot
initiate synthesis of a complementary strand of DNA on a totally
single-stranded template. Rather, they require an RNA primer, which is a short,
double-stranded region consisting of RNA base-paired to the DNA template, with
a free hydroxyl group on the 3I -end
of the RNA strand (Figure 29.15). This hydroxyl group serves as the first
acceptor of a deoxynucleotide by action of a DNA polymerase. [Note: Recall that
glycogen synthase also requires a primer.]
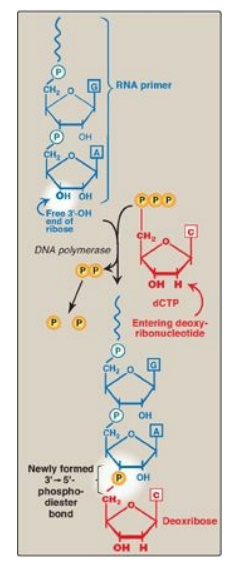
Figure 29.15 Use of an RNA primer to initiate DNA synthesis. P = phosphate.
1. Primase: A specific RNA polymerase, called primase (DnaG),
synthesizes the short stretches of RNA (approximately ten nucleotides long)
that are complementary and antiparallel to the DNA template. In the resulting
hybrid duplex, the U (uracil) in RNA pairs with A in DNA. As shown in Figure
29.16, these short RNA sequences are constantly being synthesized at the
replication fork on the lagging strand, but only one RNA sequence at the origin
of replication is required on the leading strand. The substrates for this
process are 5I -ribonucleoside triphosphates,
and pyrophosphate is released as each ribonucleoside monophosphate is added
through formation of a 3 I →5 I phosphodiester bond. [Note: The RNA
primer is later removed.]
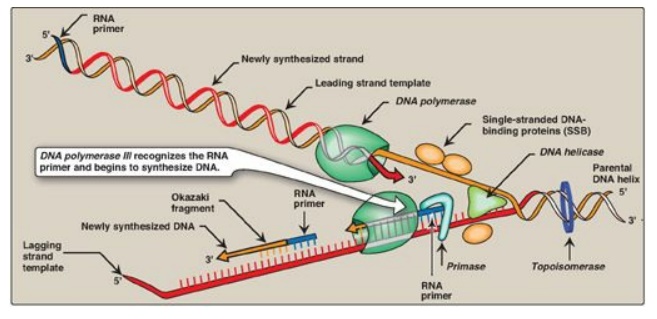
Figure 29.16 Elongation of the
leading and lagging strands. [Note: The DNA sliding clamp is not shown.]
2. Primosome: The addition of primase converts the prepriming complex
of proteins required for DNA strand separation to a primosome. The primosome
makes the RNA primer required for leading strand synthesis and initiates
Okazaki fragment formation in lagging strand synthesis. As with DNA synthesis,
the direction of synthesis of the primer is 5 →3 .
E. Chain elongation
Prokaryotic (and
eukaryotic) DNA polymerases (DNA pols) elongate a new DNA strand by adding
deoxyribonucleotides, one at a time, to the 3I-end of the growing chain (see Figure 29.16). The sequence of nucleotides
that are added is dictated by the base sequence of the template strand with
which the incoming nucleotides are paired.
1. DNA polymerase III: DNA chain elongation is catalyzed by the multisubunit enzyme, DNA polymerase III (DNA pol III). Using the 3I -hydroxyl group of the RNA primer as the acceptor of the first deoxyribonucleotide, DNA pol III begins to add nucleotides along the single-stranded template that specifies the sequence of bases in the newly synthesized chain. DNA pol III is a highly “processive” enzyme (that is, it remains bound to the template strand as it moves along and does not diffuse away and then rebind before adding each new nucleotide). The processivity of DNA pol III is the result of its β subunit forming a ring that encircles and moves along the template strand of the DNA, thus serving as a sliding DNA clamp. [Note: Clamp formation is facilitated by a protein complex, the clamp loader, and ATP hydrolysis.] The new strand grows in the 5I →3I direction, antiparallel to the parental strand (see Figure 29.16). The nucleotide substrates are 5I -deoxyribonucleoside triphosphates. Pyrophosphate (PPi) is released when each new deoxynucleoside monophosphate is added to the growing chain (see Figure 29.15). Hydrolysis of PPi to 2Pi means that a total of two high-energy bonds are used to drive the addition of each deoxynucleotide.
The production of PPi with subsequent
hydrolysis to 2P, as seen in DNA replication, is a common theme in biochemistry.
Removal of the PPi product drives a reaction in the forward
direction, making it essentially irreversible.
All four substrates
(deoxyadenosine triphosphate [dATP], deoxythymidine triphosphate [dTTP],
deoxycytidine triphosphate [dCTP], and deoxyguanosine triphosphate [dGTP]) must
be present for DNA elongation to occur. If one of the four is in short supply,
DNA synthesis stops when that nucleotide is depleted.
2. Proofreading of newly synthesized DNA: It is highly important for the
survival of an organism that the nucleotide sequence of DNA be replicated with
as few errors as possible. Misreading of the template sequence could result in
deleterious, perhaps lethal, mutations. To ensure replication fidelity, DNA pol
III has a “proofreading” activity (3I →5I exonuclease, Figure
29.17) in addition to its 5I →3I polymerase activity. As
each nucleotide is added to the chain, DNA pol III checks to make certain the
added nucleotide is, in fact, correctly matched to its complementary base on
the template. If it is not, the 3I →5I exonuclease activity
removes the error. [Note: The enzyme requires an improperly base-paired 3I
-hydroxy terminus and, therefore, does not degrade correctly paired nucleotide
sequences.] For example, if the template base is C and the enzyme mistakenly
inserts an A instead of a G into the new chain, the 3I →5I exonuclease activity
hydrolytically removes the misplaced nucleotide. The 5I →3I polymerase activity
then replaces it with the correct nucleotide containing G (see Figure 29.17).
[Note: The proofreading exonuclease activity requires movement in the 3I
→5I
direction, not 5I →3I like the polymerase activity. This is because the excision
must be done in the reverse direction from that of synthesis.]
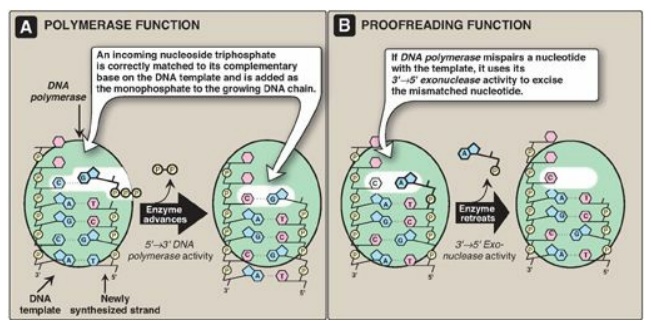
Figure 29.17 3 I →5 I Exonuclease activity enables DNA polymerase III to “proofread” the newly synthesized DNA strand.
F. Excision of RNA primers and their replacement by DNA
DNA pol III continues
to synthesize DNA on the lagging strand until it is blocked by proximity to an
RNA primer. When this occurs, the RNA is excised and the gap filled by DNA pol
I.
1. 5I →3I Exonuclease activity: In addition to having the 5 I →3 I polymerase activity that synthesizes DNA and the 3 I →5 I exonuclease activity that proofreads the newly synthesized DNA chain like DNA pol III, DNA pol I also has a 5I →3I exonuclease activity that is able to hydrolytically remove the RNA primer. [Note: These activities are exonucleases because they remove nucleotides from the end of the DNA chain, rather than cleaving the chain internally as do the endonucleases (Figure 29.18).] First, DNA pol I locates the space (nick) between the 3 I -end of the DNA newly synthesized by DNA pol III and the 5 I -end of the adjacent RNA primer. Next, DNA pol I hydrolytically removes the RNA nucleotides “ahead” of itself, moving in the 5 I →3 I direction (5 I →3 I exonuclease activity). As it removes the RNA, DNA pol I replaces it with deoxyribonucleotides, synthesizing DNA in the 5 I →3 I direction (5 I →3 I polymerase activity). As it synthesizes the DNA, it also “proofreads” the new chain using its 3I →5I exonuclease activity to remove errors. This removal/synthesis/proofreading continues, one nucleotide at a time, until the RNA primer is totally degraded, and the gap is filled with DNA (Figure 29.19). [Note: DNA pol I uses its 5I →3I polymerase activity to fill in gaps generated during DNA repair.]
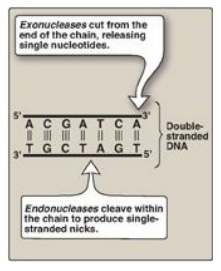
Figure 29.18 Endonuclease
versus exonuclease activity. [Note: Restriction endonucleases cleave both
strands.] T= thymine; A = adenine; C = cytosine; G = guanine.
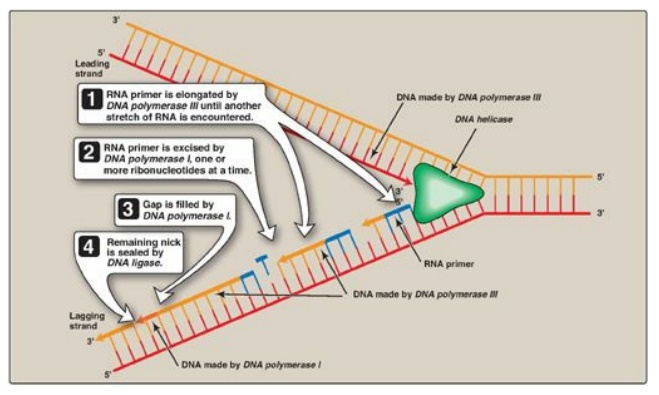
Figure 29.19 Removal of RNA primer and filling of the resulting “gaps” by DNA polymerase I.
2. Comparison of 5I →3I and 3I →5I exonucleases: The 5 I →3 I exonuclease activity of DNA pol I allows the polymerase, moving 5I →3I , to hydrolytically remove one or more nucleotides at a time from the 5 end of the 20 to 30 nucleotide–long RNA primer. In contrast, the 3 I →5 I exonuclease activity of DNA pol I, as well as DNA pol and III, allows these polymerases, moving 3 I →5 I , to hydrolytically remove one misplaced nucleotide at a time from the 3 I end of a growing DNA strand, increasing the fidelity of replication such that newly replicated DNA has one error per 107 nucleotides.
G. DNA ligase
The final
phosphodiester linkage between the 5I -phosphate group on the DNA chain
synthesized by DNA pol III and the 3I -hydroxyl group on the chain made by DNA
pol I is catalyzed by DNA ligase (Figure 29.20). The joining of these two
stretches of DNA requires energy, which in most organisms is provided by the
cleavage of ATP to AMP + PPi.
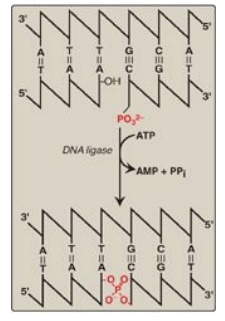
Figure 29.20 Formation of a phosphodiester bond by DNA ligase. [Note: AMP is first linked to ligase, then to the 5I phosphate, and then released.]
H. Termination
Replication termination in E. coli is mediated by sequence-specific binding of the protein, Tus (terminus utilization substance) to replication termination sites (ter sites) on the DNA, stopping the movement of DNA polymerase.
Related Topics
