Stereochemistry to Deduce Mechanism
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Stereochemical and Conformational Isomerism
In the above discussion of stereoselectivity the mechanisms of various reactions have been used to rationalize why some are stereoselective and some are not.
STEREOCHEMISTRY TO DEDUCE
MECHANISM
In
the above discussion of stereoselectivity the mechanisms of various reactions
have been used to rationalize why some are stereoselective and some are not.
Thus the bromination of olefins proceeds via a bridged bromonium ion
interme-diate and gives only trans addition across the double bond [reactions
(6.2) and (6.3)]. In contrast, the addition of HBr across a double bond gives a
carboca-tion intermediate that does not maintain the facial integrity of the
olefin and is thus much less stereoselective [reaction (6.1)]. In these
examples the mechanism of the reaction is used to explain and understand the
diastereoselectivity that is observed. There are many other examples (usually
in textbooks) where the mech-anism of a reaction is used to rationalize the
stereoselectivity of the process. To do this requires that the mechanism be
known with certainty.
In
most cases in the real world of chemistry both currently and historically, the
reverse order is followed; that is, the mechanism is deduced with certainty
only after the stereoselectivity has been determined. Because the
stereochemical out-come of a chemical reaction is experimentally determined, it
provides a powerful tool for examining the intimate spatial details of
transition states and for deter-mining how a reaction takes place at the
molecular level. As such stereochemical studies have had a huge impact on the
elucidation of reaction mechanisms.
In
addition to bond breaking and charge buildup in the transition state,
stere-ochemical changes during a reaction provide insight into the structural
require-ments of the activated complex of the rate-determining step. If a
reaction is stereospecific, that is, if only one stereoisomer is formed in a
reaction, then there is likely to be a particular spatial relationship between
groups that is required for efficient product formation. (The key here is the
term “efficient” because reactions can sometimes proceed if the correct spatial
relationship is not obtain-able, but they will go much more slowly.) If a
reaction is stereoselective, that is, if one stereoisomer is the major but not
exclusive product, then one particular spatial relationship is favored over
another in the product-forming step. If the stereoselectivity of a reaction can
be understood, then key structural elements in the activated complex can often
be identified.
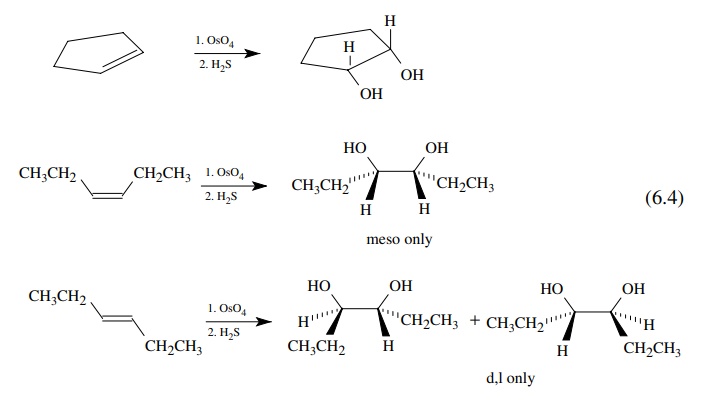
Several
common examples show the power of this reasoning. The reaction of osmium
tetroxide with olefins followed by reduction gives diols resulting exclu-sively
from syn addition to the double bond. The reaction is hence stereospecific The
addition stereochemistry is clearly seen in cyclic olefins, but it is also seen
in acyclic olefins where single diastereomers are produced [reaction (6.4)].
The
results from the cyclic series show that both oxygens come from the same side
of the double bond, probably from a single OsO4 molecule. The
results in the acyclic series demonstrate that both oxygens add to the ends of
the double bond at the same time. If one oxygen added first, an intermediate
with a sin-gle carbon – carbon bond would be formed which could isomerize by
rotation around that bond. The observation of complete diastereoselectivity
requires that both C–O bonds be formed simultaneously. Thus a concerted
addition across the double bond is the most reasonable pathway consistent with
these results. The stereochemical analysis is shown below for the cis starting
material.
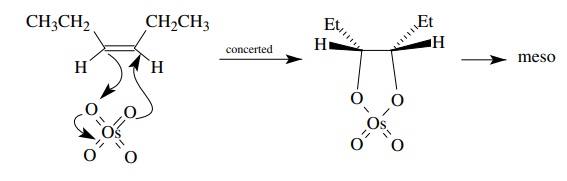
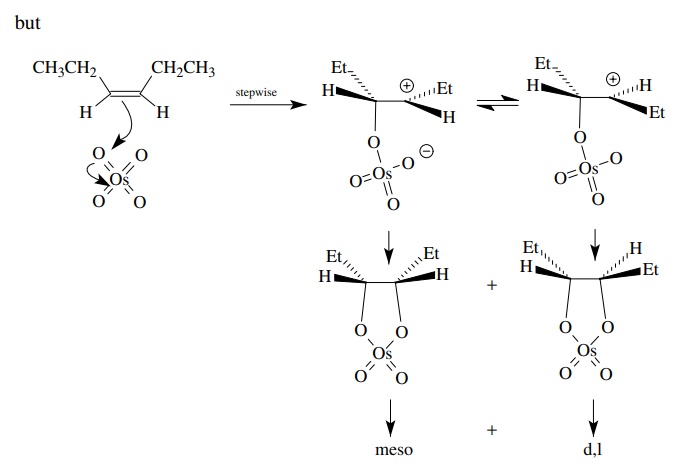
An
analogous analysis for the trans olefin would predict only the d,l diastereomer
for concerted addition of OsO4 to the π bond, but the same meso – d,l mixture would be obtained if the
addition were stepwise.
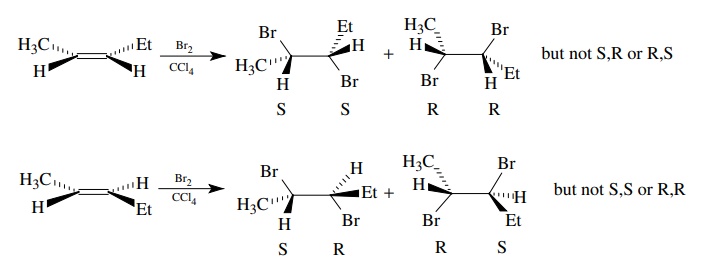
Contrast
the above syn addition of osmium tetroxide with the well-known anti
stereochemistry found in the addition of bromine to alkenes. Cyclic systems
give only trans addition in most cases, and acyclic olefins give single
diastereomers that depend on the geometry of the starting olefin. These results
are consistent with one bromine adding to one face of the olefin to give a
bridged ion which maintains the stereochemistry of the original olefin. Bromide
ion adds from the opposite face to give a single diastereomeric dibromide
product.
Walden inversion was the term given
to the change in stereochemistry observed
in bimolecular nucleophilic substitutions. For example, reaction of (2S)-2-triflyloxyesters with sodium azide
gives (2R)-2-azidoesters.
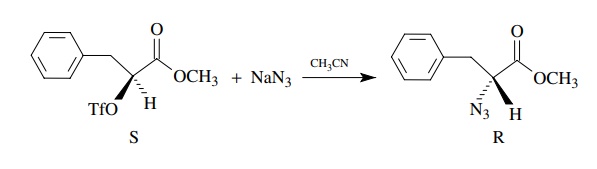
Inversion
of configuration requires that the nucleophile adds electrons to the σ ∗ orbital of the
carbon – triflate bond from the side opposite that bond. As required by the
stereochemistry, formation of the bond from azide to carbon is concurrent with
cleavage of the carbon leaving group bond.
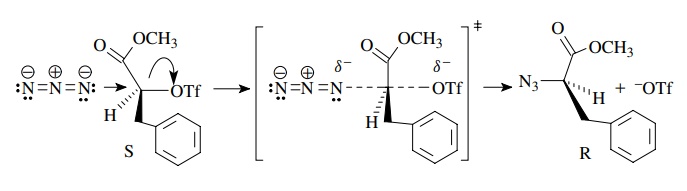
If
racemization were observed, it could only be due to a cleavage of one of the
bonds to the chiral center prior to carbon – nitrogen bond formation or
subsequent to it. This could occur by (a) enolization of the starting triflate,
(b) an ionization of triflate to a carbocation and then nucleophilic attack by
the azide, or (c) enolization in the azido product. The fact that clean
inversion occurs means not only that the substitution by azide occurs with
inversion but also that none of these other processes is significant under the
reaction conditions since they would lead to racemized product.
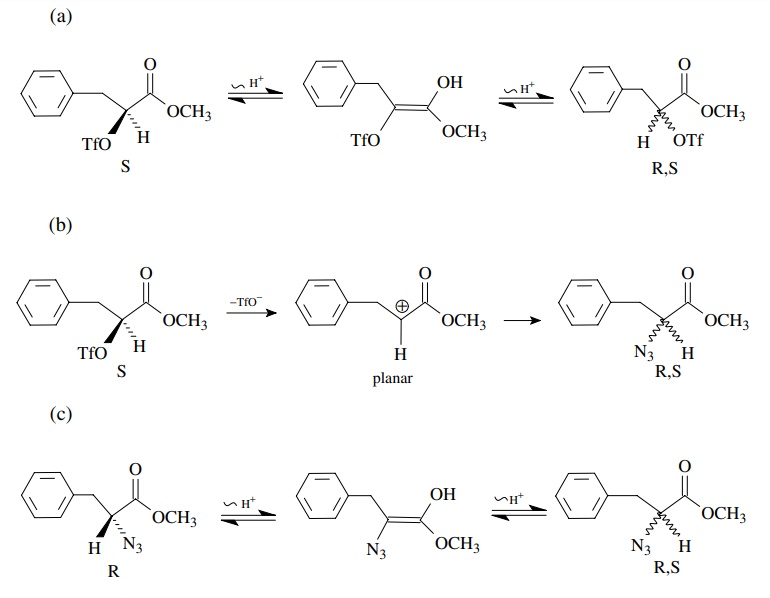
The
stereoelectronic requirements of groups undergoing base-promoted elimination is
also easily seen by stereochemical studies. Treatment of trans-2-methylcyclohexyl tosylate gives 3-methylcyclohexene as the
major product while treatment of cis-2-methylcyclohexyl
tosylate gives the more stable 1-methylcyclohexene as the only product.
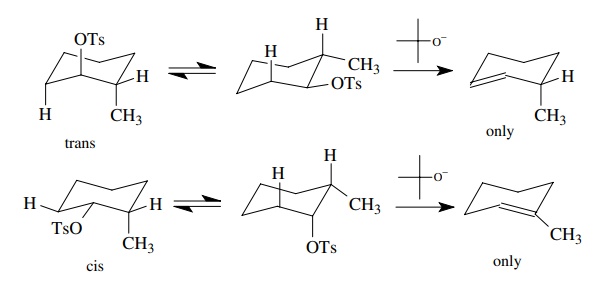
These
data are consistent with the favored transition state having an antiperiplanar
relationship between the proton being removed and the leaving group. In a
six-membered ring, this can only occur when they both are diaxial. In the trans
isomer, the conformation in which the tosylate is axial only has a proton at
C-6 axial and antiperiplanar. Thus elimination occurs across C-6 and C-1 to
give only 3-methyl cyclohexene. In the cis isomer, the conformation in which
the tosylate group is axial has antiperiplanar hydrogens at both C-2 and C-6. Elimination could
proceed in either direction; however, removal of the proton at C-2 is favored
because the more stable olefin product is produced.
These
examples show the power of stereochemical information in pinpointing structural
elements of activated complexes. Combined with other types of mech-anistic
information, even the most intimate mechanistic details can be clarified in
many cases. For example, consider the solvolysis in ethanol of
3-phenyl-2-tosyloxy butane in which the replacement of the tosylate group by a
solvent nucleophile is noted.
While
this appears to be a simple substitution reaction, the details can be further
explored. It was found that this reaction proceeded by a first-order rate law,
which suggests an ionization pathway (Sn1) for the substitution. However, when
a group of substituted aromatic compounds were investigated, plots of the rate
constants (log kZ/ kH) gave a much better
correlation with σZ+ than with σz and ρ+ = −1.3. This Hammett study reveals that a
positive charge is developed on the aromatic ring in the transition state of
the rate-determining step. About the only way for this to happen is for the
phenyl ring to interact with the positive charge produced by the ionization of
the leaving group.
The
use of the 2R,3R isomer led to formation of only 2R,3R-2-ethoxy-3-phenylbutane.
Thus the configuration at each chiral center was retained in the product. These
stereochemical data rule out simple ionization and solvent capture as a
reaction mechanism since this would lead to a mixture of 2R and 2S
configurations. From these observations it has been postulated that the phenyl
group assists ionization of the leaving group by electron donation to produce a
bridged ion.
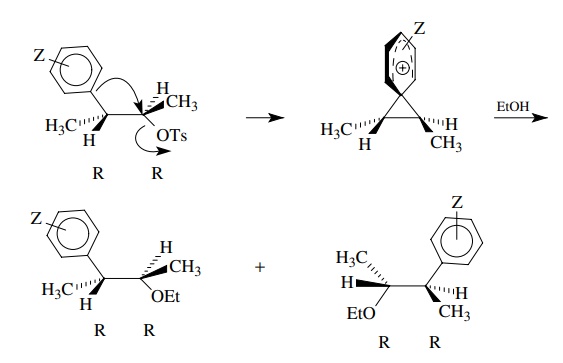
The
bridged ion has a positive charge delocalized over the aromatic ring as
required by the σZ+ correlation.
Furthermore the solvent nucleophile can only add from the side opposite the
bridging phenyl group, leading to retention of configuration as the
stereochemical results demand.
Stereochemical
studies can be an indispensable adjunct to other types of mech-anistic
investigations for unraveling the details of reaction processes. They allow the
positions of atoms or groups in a molecule to be tracked through a reaction,
thereby revealing the spatial requirements of the reaction.
Related Topics
