Type 2 Diabetes
| Home | | Biochemistry |Chapter: Biochemistry : Diabetes Mellitus
T2D is the most common form of the disease, afflicting over 90% of the diabetic population in the United States.
TYPE 2 DIABETES
T2D is the most common
form of the disease, afflicting over 90% of the diabetic population in the
United States. [Note: American Indians, Alaskan Natives, Hispanic and Latino
Americans, African Americans, and Asian Americans have the highest prevalence.]
Typically, T2D develops gradually without obvious symptoms. The disease is
often detected by routine screening tests. However, many individuals with T2D
have symptoms of polyuria and polydipsia of several weeks’ duration. Polyphagia
may be present but is less common. Patients with T2D have a combination of
insulin resistance and dysfunctional β cells (Figure 25.6) but do not require
insulin to sustain life, although insulin eventually will be required to
control hyperglycemia and keep HbA1c below 7% in over 90% of patients. The
metabolic alterations observed in T2D are milder than those described for type
1, in part, because insulin secretion in T2D, although inadequate, does
restrain ketogenesis and blunts the development of DKA. (Recall that insulin
suppresses the release of glucagon.) Diagnosis is based on the presence of
hyperglycemia as described above. The pathogenesis does not involve viruses or
autoimmune antibodies and is not completely understood. [Note: An acute
complication of T2D in the elderly is a hyperosmolar hyperglycemic state
characterized by severe hyperglycemia and dehydration and altered mental
status.]
T2D is characterized by hyperglycemia; insulin
resistance; impaired insulin secretion; and, ultimately, β-cell failure. The
eventual need for insulin therapy has eliminated the designation of T2D as
“noninsulin-dependent” diabetes.
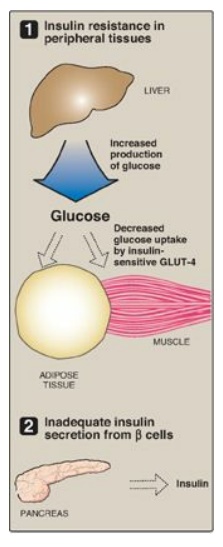
Figure 25.6 Major factors
contributing to hyperglycemia observed in type 2 diabetes. GLUT = glucose
transporter.
A. Insulin resistance
Insulin resistance is
the decreased ability of target tissues, such as liver, adipose, and muscle, to
respond properly to normal (or elevated) circulating concentrations of insulin.
For example, insulin resistance is characterized by increased hepatic glucose
production, decreased glucose uptake by muscle and adipose tissue, and
increased adipose lipolysis with production of free fatty acids ([FFAs] Figure
25.7).
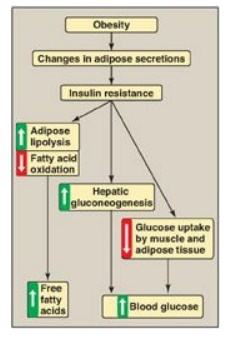
Figure 25.7 Obesity, insulin resistance, and hyperglycemia. [Note: Inflammation also is associated with insulin resistance.]
1. Insulin resistance and obesity: Although obesity is the most common cause of insulin resistance and T2D, most people with obesity and insulin resistance do not become diabetic. In the absence of a defect in β-cell function, nondiabetic, obese individuals can compensate for insulin resistance with elevated levels of insulin. For example, Figure 25.8A shows that insulin secretion is two to three times higher in obese subjects than it is in lean individuals. This higher insulin concentration compensates for the diminished effect of the hormone (as a result of insulin resistance) and produces blood glucose levels similar to those observed in lean individuals (Figure 25.8B).
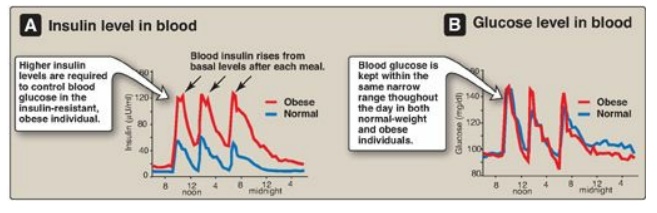
Figure 25.8 Blood insulin and glucose levels in normal-weight and obese subjects.
2. Insulin resistance and type 2 diabetes: Insulin resistance alone will not lead to T2D. Rather, T2D develops in insulin-resistant individuals who also show impaired β-cell function. Insulin resistance and subsequent risk for the development of T2D is commonly observed in individuals who are obese, physically inactive, or elderly and in the 3%–5% of pregnant women who develop gestational diabetes. These patients are unable to sufficiently compensate for insulin resistance with increased insulin release. Figure 25.9 shows the time course for the development of hyperglycemia and the loss of β-cell function.
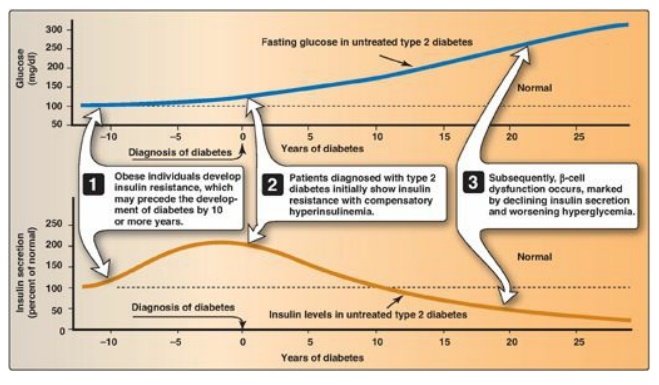
Figure 25.9 Progression of blood glucose and insulin levels in patients with type 2 diabetes.
3. Causes of insulin resistance: Insulin resistance increases with weight gain and decreases with weight loss, and excess adipose tissue is key in the development of insulin resistance (see Figure 25.7). Adipose tissue is not simply an energy storage organ, but also a secretory organ. With obesity, there are changes in adipose secretions that result in insulin resistance. These include secretion of proinflammatory cytokines such as interleukin 6 (inflammation is associated with insulin resistance); increased synthesis of leptin, a protein with proinflammatory effects; and decreased secretion of adiponectin, an adipocyte protein with anti-inflammatory effects. One effect of insulin resistance is increased lipolysis and production of FFAs. FFA availability decreases use of glucose, contributing to hyperglycemia, and increases deposition of TAG in liver (hepatic steatosis). FFAs also have a proinflammatory effect. In the long-term, FFAs suppress glucose-induced insulin release. [Note: Adiponectin increases FA β-oxidation. Consequently, a decrease in this adipocyte protein contributes to FFA availability.]
B. Dysfunctional β cells
In T2D, the pancreas
initially retains β-cell capacity, resulting in insulin levels that vary from
above normal to below normal. However, with time, the β cell becomes increasingly
dysfunctional and fails to secrete enough insulin to correct the prevailing
hyperglycemia. For example, insulin levels are high in typical, obese, T2D
patients but not as high as in similarly obese individuals who are nondiabetic.
Thus, the natural progression of the disease results in a declining ability to
control hyperglycemia with endogenous secretion of insulin (Figure 25.10).
Deterioration of β-cell function may be accelerated by the toxic effects of
sustained hyperglycemia and elevated FFAs and a proinflammatory environment.
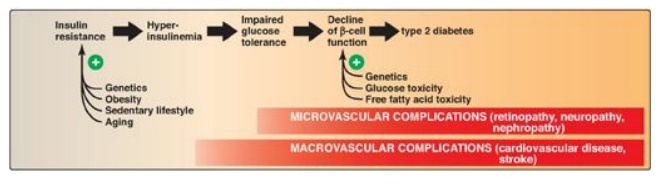
Figure 25.10 Typical
progression of type 2 diabetes.
C. Metabolic changes in type 2 diabetes
The metabolic
abnormalities of T2D are the result of insulin resistance expressed primarily
in liver, muscle, and adipose tissue (Figure 25.11).
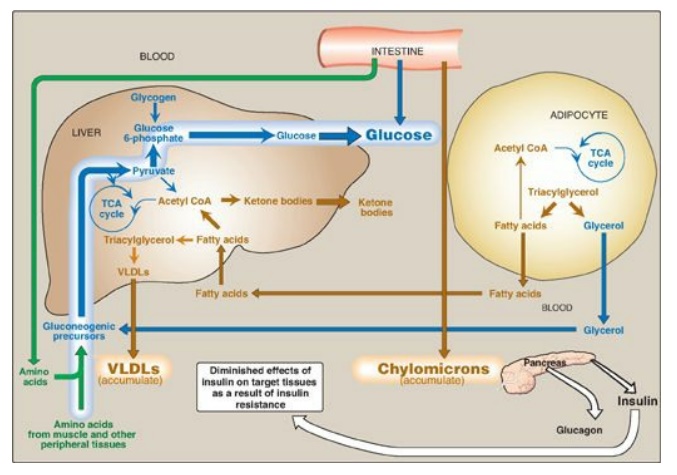
Figure 25.11 Intertissue relationships in type 2 diabetes. [Note: Ketogenesis is restrained as long as insulin action is adequate.] TCA = tricarboxylic acid; CoA = coenzyme A; VLDL = very-low-density lipoprotein.
1. Hyperglycemia: Hyperglycemia is caused by increased hepatic
production of glucose, combined with diminished peripheral use. Ketosis is
usually minimal or absent in patients with T2D because the presence of insulin,
even in the presence of insulin resistance, restrains hepatic ketogenesis.
2. Dyslipidemia: In the liver, FAs are converted to TAGs, which are
packaged and secreted in VLDL. Chylomicrons are synthesized from dietary lipids
by the intestinal mucosal cells following a meal. Because lipoprotein
degradation catalyzed by lipoprotein lipase in adipose tissue is low in
diabetics, the plasma chylomicron and VLDL levels are elevated, resulting in
hypertriacylglycerolemia (see Figure 25.10). Low levels of high-density
lipoproteins are also associated with T2D, likely as a result of increased
degradation.
D. Treatment of type 2 diabetes
The goal in treating T2D is to maintain blood glucose concentrations within normal limits and to prevent the development of long-term complications. Weight reduction, exercise, and medical nutrition therapy (dietary modifications) often correct the hyperglycemia of newly diagnosed T2D. Hypoglycemic agents (for example, metformin, which decreases hepatic output of glucose), sulfonylureas (increase insulin secretion;), thiazolidinediones (increase peripheral insulin sensitivity), α-glucosidase inhibitors (decrease absorption of dietary carbohydrate) or insulin therapy may be required to achieve satisfactory plasma glucose levels. [Note: Bariatric surgery in morbidly obese individuals with T2D has been shown to result in disease remission in most patients. Remission may not be permanent.]
Related Topics
