Conformational Analysis
| Home | | Organic Chemistry |Chapter: Organic Chemistry : Stereochemical and Conformational Isomerism
In the most basic sense chemical reactions are really only changes in the distribution of electrons. Such changes result in the breaking and making of chemical bonds and cause reactants to be converted to products.
CONFORMATIONAL ANALYSIS
In
the most basic sense chemical reactions are really only changes in the distribution
of electrons. Such changes result in the breaking and making of chemical bonds
and cause reactants to be converted to products. However, before such
electronic changes can take place, molecules taking part in the reaction must
approach each other within bonding distance or they must undergo a change in
geometry which permits overlap between the necessary orbitals to take place
which results in electron redistribution. Restated another way, for chemistry
to occur, molecules must first interact in a spatial sense. Consequently the
shapes of molecules and the surface features they display are an important
influence on their interactions with other molecules.
The
large number of σ bonds present in
organic molecules has a direct bearing on their shapes. Since a σ bond is axially symmetric along the
bond, rotations of the groups connected by a σ bond do not cause it to break. (Such cannot be said of π bonds!) Thus molecules with many σ bonds are capable of large numbers of
internal rotational motions which largely determine the shape, size, and
surface characteristics of the molecule. Conformational analysis is the study
of rotational motions in molecules and how they affect molecular properties.
The
simplest hydrocarbon capable of internal rotational motion is ethane. Ethane
has two tetrahedral methyl groups connected by a carbon – carbon σ bond. As such the methyl groups are
free to rotate one relative to the other. However, it is found that the various
rotational positions are not equivalent spatially or energetically. In fact,
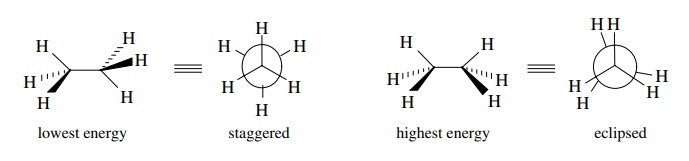
there are two limiting rotational positions for ethane. The lowest energy
conformation (synonymous with a conformational isomer) is the one in which the
C–H bonds of each methyl group are staggered between the C–H bonds of the other
methyl group across the σ bond. This
is the lowest energy conformation because the electron clouds of the bonds are
the farthest distance apart, and their repulsions are minimized.
The
highest energy conformation of ethane is the one in which the C–H bonds of each
methyl groups are eclipsed with the C–H bonds of the other methyl group across
the σ bond. This is the highest
energy because the electron clouds of the C–H bonds are as close as they can
be, and their repulsions raise the energy of the molecule.
The
staggered and eclipsed forms of ethane are conformational stereoisomers
(conformational isomers, conformers) because they have the same molecular
for-mulas and sequences of bonded elements but different spatial arrangements
due to rotations around single bonds. (Actually there are an infinite number of
con-formational isomers (also called conformations) because there are an
infinite number of degrees of rotation around the bond, but normally one only
needs to be concerned with energy minima and maxima.)
The
difference in energy between the higher and lower energy forms of ethane is
only 2.9 kcal/mol (12 kJ/mol); thus rotations around the bond are very rapid at
However, if one
plots the change in energy as ethane rotates between the staggered and eclipsed
forms, a periodic behavior is seen (Figure 6.5). Moreover, if a large number of
snapshots of ethane were taken, they would show that most of the time ethane is
found in the staggered conformation. The equilibrium between the staggered and
eclipsed conformations favors the staggered by 99.2% to 0.8%.
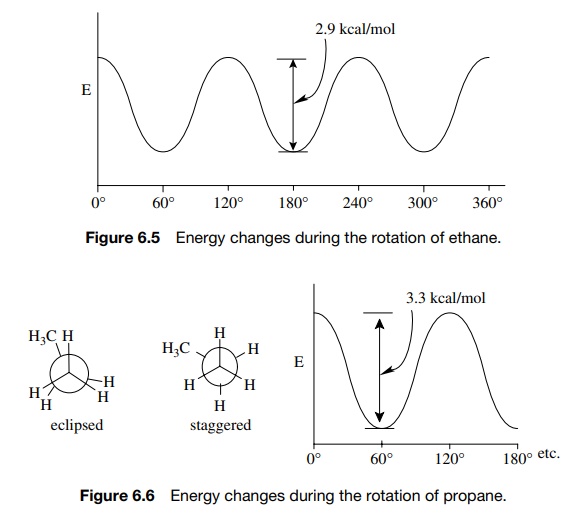
A
similar analysis of propane reveals analogous behavior with two major
conformations—staggered and eclipsed —and periodic energy changes as rota-tion
about a sigma bond occurs (Figure 6.6). There is a difference from ethane,
however, in that the energy difference between the staggered and eclipsed
con-formations is now 3.3 kcal/mol (14 kJ/mol). This increase means that a
hydrogen and methyl group eclipsed across a carbon – carbon bond repel each
other more than two hydrogen atoms. This suggests that the electron cloud of
the methyl group comes closer to the electron cloud of the C–H bond so the
repulsion is greater. Since the electron clouds associated with the methyl
group define the space that the methyl group occupies, it is clear that a
methyl group occupies more space than a hydrogen, that is, it is “larger.”
Thus, it follows that groups in molecules have definite sizes, and the size of
these groups is one factor which contributes to the overall shape of the
molecule because of its influence on the preferred conformation of the
molecule.
Conformational isomerism around the central bond in butane is more com-plex because the various staggered and eclipsed conformations are not equiva-lent as they are in ethane and propane (Figure 6.7).
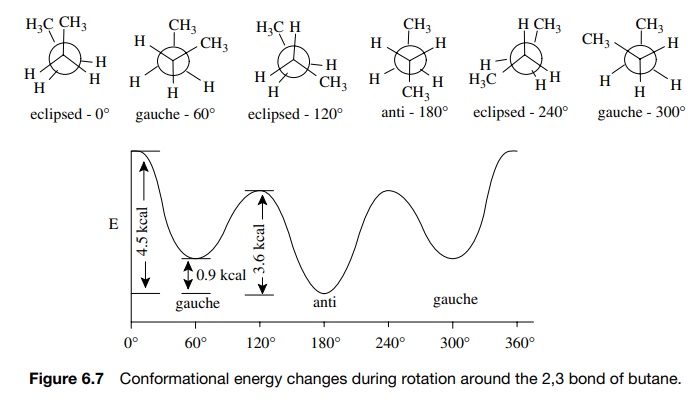
Starting with the eclipsed conformation
with the dihedral angle between the two methyl groups at 0◦ , rota-tion around
the central bond leads to two different staggered conformations and one
additional eclipsed conformation.
The
most stable staggered conformation is that in which the methyl groups are
antiperiplanar (dihedral angle of 180◦ ) —called the anti conformation.
The other staggered conformation is that in which the dihedral angle between
the methyl groups is 60◦
—called the gauche conformation (there are two of them for rotations of 60◦ or 300◦ , respectively).
The other eclipsed conformation is that in which the two methyl groups each
eclipse a hydrogen (there are two of them for rotations of +120◦ and 240◦ , respectively).
From
the energy diagram it is seen that the gauche conformation is 0.9 kcal/mol (3.7
kJ/mol) higher than the anticonformation. This must be due to some residual
repulsion between the methyl groups when the dihedral angle is only 60◦ between them. Also
the energy difference between the anti conformer and the highest energy
eclipsed conformer is 4.5 kcal/mol (18.8 kJ/mol). Thus the greater effective
size of the methyl groups results in increased repulsion when they are
eclipsed.
In addition to conformational isomerism about the 2,3 bond in butane, rotations about the 1,2 bond and the 3,4 bond are possible. The energy changes here are much smaller and are comparable to those found in propane.
The
importance of conformational isomerism lies in the fact that the predom-inant
shape that molecules adopt is dependent on the energies of the various
staggered and eclipsed conformations. In combination they can be used to
pre-dict the probable shapes the molecule normally assumes, and these shapes
are those which are presented to reagents in solution.
In
contrast to open-chain systems in which groups can rotate through 360◦ around σ bonds, cyclic systems can undergo
conformational change through only limited ranges. Like open-chain systems,
however, conformational changes in rings minimize eclipsing interactions across
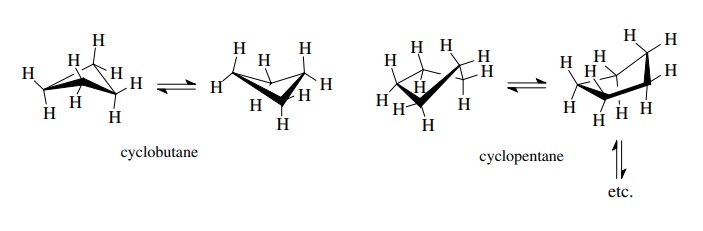
σ bonds. Cyclopropane is a flat ring
without conformational motion. Cyclobutane is not planar because, if it were,
all the C–H bonds around the ring would be eclipsed. The molecule undergoes a
conformational change that bends the molecule out of planarity by about 35◦ . This reduces
eclipsing and leads to a lower overall energy. A similar situation is found in
cyclopentane, which adopts an envelope conformation (one ring apex out of
plane) which is in equilibrium with four other envelope conformations (each
apex up) to avoid the 10 C–H eclipsing interactions that would be present if
the molecule were planar.
Saturated
six-membered rings are the most common ring systems in nature because they
present an optimal conformational situation. As seen in cyclohexane, the
molecule adopts a puckered shape called a chair
conformation in order to avoid angle strain in the ring bonds. In the chair
form, all bond angles are 109◦ and
all the bonds are staggered.
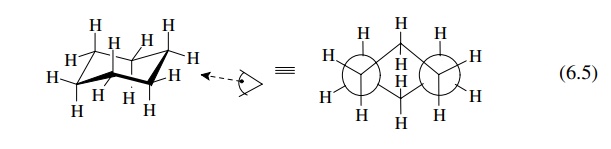
This
results in a molecule whose energy is comparable to a completely stag-gered,
open-chain alkane. This is easy to see by viewing the molecule in a Newman
projection. Viewing the molecule through the ring so that the two side carbon –
carbon bonds are seen head on, as in a Newman projection, generates the view in
(6.5).
The
chair conformer can undergo conformational isomerism to a second chair
conformer which is degenerate in energy with the first. Cyclohexane is thus a
dynamic molecule which exists largely in one of two chair isomers. These are
the lowest energy conformations. Other higher energy conformations of
cyclohexane include the boat form, which is 10.1 kcal/mol (42.3 kJ/mol) above
the chair form, and the twist boat form, which lies 3.8 kcal/mol (15.9 kJ/mol)
above the chair form.
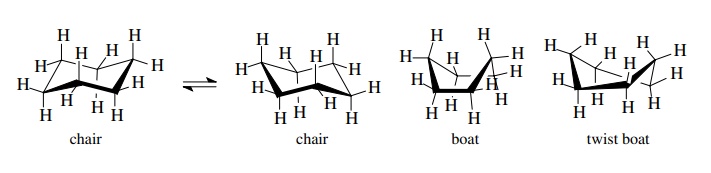
Although
these are well-defined conformational isomers, their energies are such that
they are virtually unpopulated at room temperature. (The twist boat is an
intermediate in the conversion of one chair form to the other.) At the same
time the conversion of one chair form to the other occurs rapidly at room
temperature, and both chair forms are in rapid equilibrium.
Because
cyclohexane exists in the chair form, the C–H bonds of the methylene groups are
nonequivalent. There are two types of valences on each CH2 group.
One type is perpendicular to a plane loosely defined by the ring carbons and is
called axial. The second type falls generally in the plane loosely defined by
the ring carbons and is termed equatorial. These are shown both in combination
and individually.
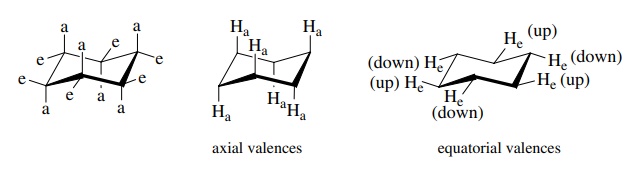
Three
axial valences on alternate (1,3) carbons point to one side of the ring (up)
and the other three axial valences on alternate (1,3) carbons point to the
other side of the ring (down). The same is true for equatorial valences; while
the directionality is not so obvious for equatorial valences; they still point
toward one side of the ring (up) or the other (down).
There
are two chair forms and two types of valences (axial and equatorial). The
conversion of one chair form to the other interconverts the axial and
equatorial valences (i.e., a valence which is axial in one chair form is
equatorial in the other chair form and vice versa). In the structures below one
of the carbons is indexed with a star ( *) to help keep track of it.
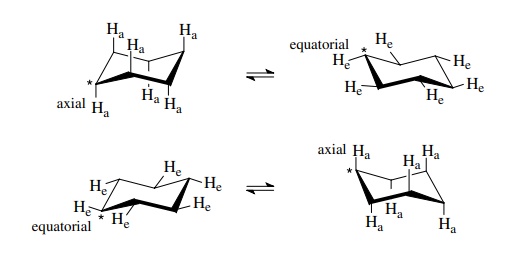
In
cyclohexane the chair forms have equal energy, but if groups other than
hydrogen are attached to the cyclohexane ring, the two chair forms are no
longer equivalent. In one chair isomer the group is equatorial and in the other
chair isomer it must be axial. This is shown for methylcyclohexane.
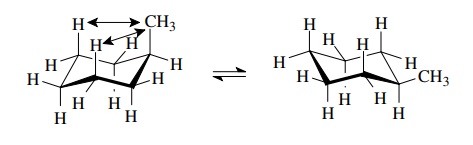
Both
conformations have the methyl group staggered between the vicinal pro-tons.
When the methyl group is axial, it is sufficiently close to the syn – axial
protons to undergo 1,3 diaxial interactions and be repelled by them. This
raises the energy of the axial conformer relative to the equatorial conformer.
For a methyl group, the energy difference is about 1.8 kcal/mol. (Actually, the
rela-tionship of an axial methyl group to the ring bonds is a gauche
conformational relationship. Thus the value of 1.8 kcal/mol for an axial methyl
group is the value of two gauche butane interactions with the ring bonds!)
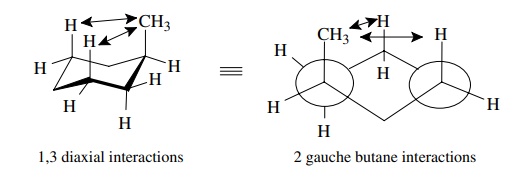
Other
groups would behave similarly, with the axial isomer being higher in energy
(less stable) than the equatorial isomer because of 1,3 diaxial interactions.
These two isomers are conformational isomers because they are interconvertible
by rotations about C–C single bonds, but they are also called conformational
diastereomers since they have different physical properties and are
nonsuperim-posable, non-mirror images.
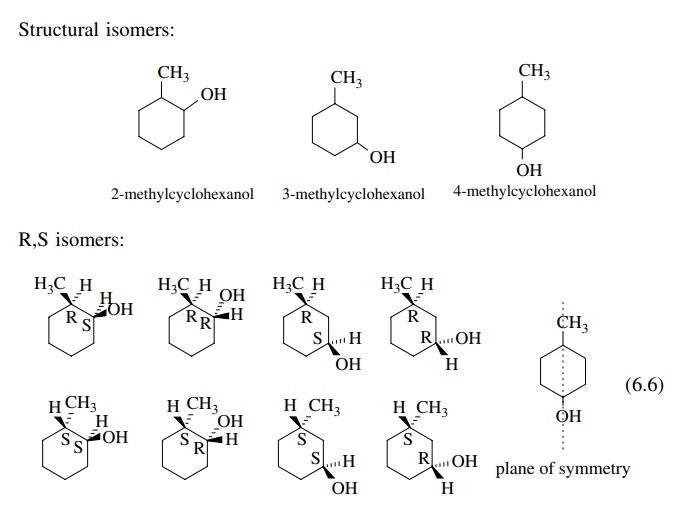
When
more than one group is attached to cyclohexane, the stereoisomeric
pos-sibilities increase. First, structural isomers of the 1,2, 1,3, or 1,4 type
are possible.
Next
relative configurations (R,S) are possible for 1,2- or 1,3-disubstituted
isomers. (The 1,4 isomer has a plane of symmetry.) The relative stereochemistry
can be denoted as cis or trans, depending on whether the substituents point
toward the same side or opposite sides of the ring. Finally, the cyclohexane
ring can undergo chair – chair interconversion leading to different
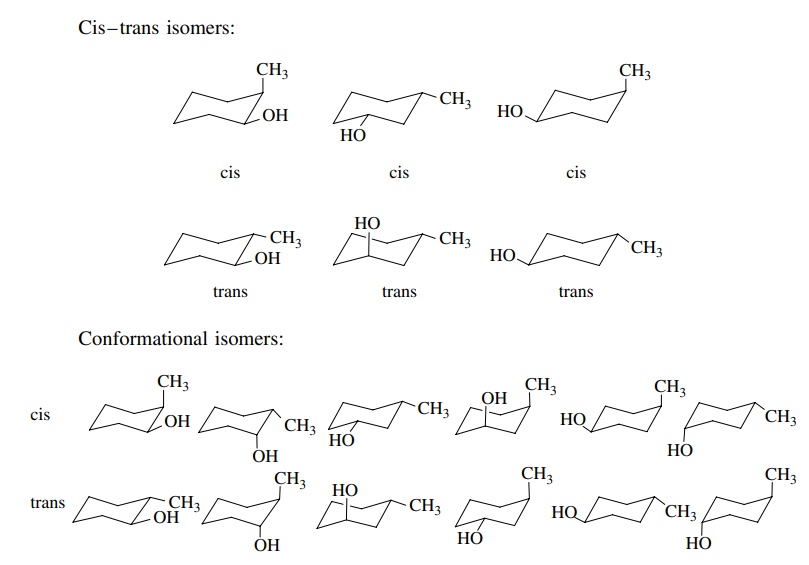
conformational isomers. These possibilities are shown for methylcyclohexanol in
(6.6).
The
first three types of isomerism are familiar and have been discussed
previ-ously. The conformational isomerism is very understandable if it is
remembered that axial and equatorial valences exchange upon chair – chair
interconversion. For example, to draw the trans isomer of 3-methylcyclohexanol,
one of the groups must be equatorial and the other axial. The other chair form must have the groups in opposite
valences. Similarly trans-2-methyl-cyclohexanol
has both groups equatorial in one chair form. The other chair form must
therefore have both groups axial.
Related Topics
