Cephalosporins
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Antibiotics And Synthetic Antimicrobial Agents: Their Properties And Uses
There are quite a number of similarities between cephalosporins and penicillins, including their early history. In both cases, the academic research was conducted at Oxford University and the American pharmaceutical companies converted that early work into a marketed product.
CEPHALOSPORINS
There are quite a number of similarities between cephalosporins and penicillins,
including their early history. In both cases, the academic research was
conducted at Oxford University and the American pharmaceutical companies
converted that early work into a marketed product. The discovery of the first
cephalosporin was made in 1948 in Sardinia, the research was conducted at the
William Dunn School of Pathology during the 1950s and the first antibiotics in
the class, cephalothin and cephaloridine, became available in the mid 1960s.
Since then many cephalosporins have been synthesized, and more than 60 have
been marketed in various countries over the last 40 years, although not all of
them are still available. As with the penicillins, the policy in this chapter
is to consider the general properties of this class of antibiotics and to
provide more detail about selected important cephalosporins, rather than
provide a comprehensive listing.
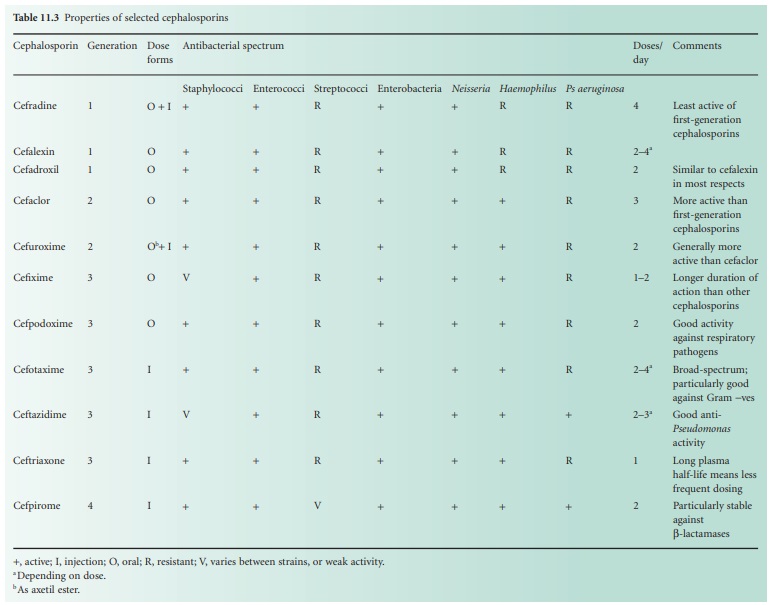
Cephalosporins consist of a
six-membered dihydrothiazine ring fused to a β-lactam ring (top structure
in Figure 11.4).
The position of the double bond in Δ3-cephalosporins
is important, since Δ2-cephalosporins (double bond
between 2 and 3) are not antibacterial, irrespective of the composition of the
side chains. The similarity to the basic penicillin structure is immediately
apparent, but the crucial difference is that there is much greater scope for
structural modification of the cephalosporins because of the presence of two
side chains (on carbons 3 and 7) in contrast to the single side chain of
penicillins. Again, the fundamental properties of acid stability (and hence
oral availability), antimicrobial spectrum, resistance to β-lactamases and
pharmacokinetics can all be substantially varied by sidechain modifications.
Several of the cephalosporins act as good inducers of β-lactamases.
The many cephalosporins have been classified into ‘g enerations’,
although the usefulness of such a classification has been questioned. There is
general acceptance of four generations, but ceftobiprole has been claimed as
the first member of the fifth. The assignment of cephalosporins into the first
four generations has broadly followed the time course of their introduction,
but there has been some overlap, so certain drugs that have been classified
into one particular generation were marketed in some countries after the
earliest ones of the next. The situation is further complicated by the fact
that there is not universal agreement on the generation to which some
cephalosporins belong; cefaclor, for example, is considered a first-generation
antibiotic in Japan, but is regarded as second-generation in most other
countries.
The general trends have been for an
increase in activity towards Gramnegative species (usually with a corresponding
loss of anti-staphylococcal action), and increased resistance to β-lactamase as
the development of cephalosporins has progressed through the generations.
First-generation drugs are those that have moderate antimicrobial activity and
resistance to staphylococcal, but not Gram-negative, β-lactamases. These were
originally used primarily as alternative antibiotics for the treatment of
staphylococcal infections, and are rarely first-choice therapy. Like the oral
penicillins, the oral cephalosporins may disturb the gut flora and give rise to
diarrhoea. The possession of good resistance to both staphylococcal and
Gram-negative β-lactamases is the principal characteristic distinguishing the
second from the first-generation antibiotics, although improved potency,
particularly towards H. influenzae and
enterobacteria, is also a feature. Yet higher activity towards Gram-negative
bacteria is displayed by third generation drugs, to an extent that some of them
have little or no value in the treatment of staphylococcal infections. The
parenterally administered third generation cephalosporins, e.g. cefotaxime and
ceftazidime, are sometimes used in combination with gentamicin or other
aminoglycosides with the intention of achieving synergy. The usefulness of the
third generation drugs has diminished somewhat since their introduction as a consequence
of the spread of strains capable of producing extended-spectrum β-lactamases
and it was this deficiency that the fourth-generation cephalosporins were
intended to remedy. Cefpirome and cefepime exhibit extremely good enzyme
resistance, but otherwise have much the same antibacterial spectrum as
ceftazidime and other third-generation molecules.
Structure-activity relationships
The activity of cephalosporins (and
other β-lactams) against Gram-positive bacteria depends on antibiotic affinity for
penicillin-sensitive enzymes (PSEs) also known as penicillin binding proteins
(PBPs). Resistance results from altered PBPs or, more commonly, from
β-lactamases. Activity against Gram-negative bacteria depends on penetration of
β-lactams through the outer membrane, resistance to β-lactamases found in the
periplasmic space and binding to PBPs. (For further information on mechanisms
of action and bacterial resistance, see Chapters 12 and 13). Modification of
the cephalosporin nucleus (Figure 11.4)
at 7α (i.e. R3) by addition of a methoxy group
increases β-lactamase stability but decreases activity against Gram-positive
bacteria because of reduced affinity for PBPs; molecules possessing a
7α-methoxy group, e.g. cefoxitin, are termed cephamycins.
Side chains containing a
2-aminothiazolyl group at R1, e.g.
cefotaxime, ceftriaxone and ceftazidime, yield cephalosporins with enhanced
affinity for PBPs of Gram-negative bacteria and streptococci. An iminomethoxy
group (-C=N.OCH3) in, for example, cefuroxime,
provides β-lactamase stability against common plasmid-mediated β-lactamases. A
propylcarboxy group ((CH3)2-C-COOH) as in ceftazidime increases β-lactamase
resistance and also provides activity against Ps. aeruginosa,
while at the same time reducing β-lactamase induction capabilities. In
cephalosporins susceptible to β-lactamases, opening of the β-lactam ring occurs
with concomitant loss of the substituent at R2 (except in
cefalexin, where R2 represents H; see Figure 11.4).
This is followed by fragmentation of the molecule.
The nature of the R2 substituent influences both the
pharmacokinetic properties of the molecule and its ability to enter bacterial
cells—particularly to cross the outer membrane of Gram-negative bacteria via
porins. For good oral absorption: (1) the R2 substituent
must be small, non-polar and stable; a methyl group is considered desirable but
might decrease antibacterial activity; and (2) the 7-acyl group (R1) must be based on phenylglycine and the amino group
must remain unsubstituted. Esterification of the carboxylic acid group at C4
can, as with the penicillins, result in enhanced oral absorption provided that
the ester is rapidly hydrolysed by tissue esterases; this is exemplified in
both cefuroxime axetil and cefpodoxime proxetil. The possession of a quaternary
nitrogen on the side chain at position 3 has two benefits: it reduces the
affinity of the cephalosporin for Gram-negative β-lactamases, in other words,
makes it more resistant to enzyme attack, and it makes the molecule zwitterionic
which increases the rate at which it can pass through the porin channels into
the Gram-negative cell.
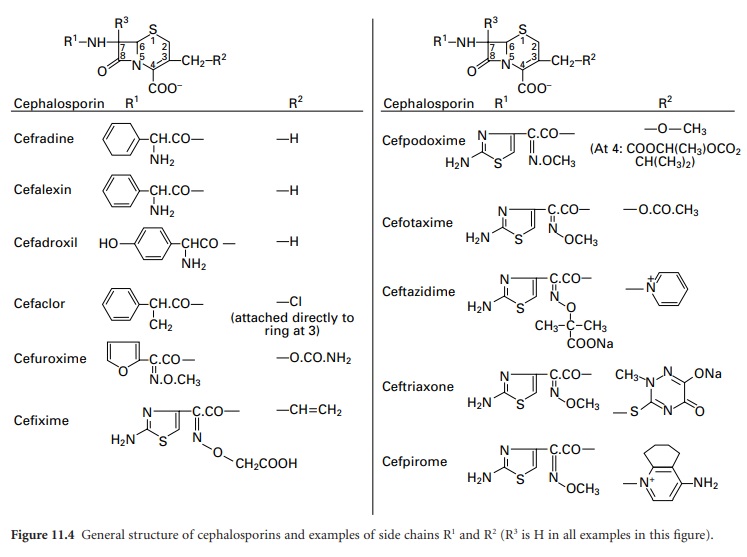
Side chains of the various cephalosporins,
including those most recently developed, are presented in Figure 11.4 and
a summary of the properties of these antibiotics in Table 11.3.