Chapter Summary, Questions Answers - Glycogen Metabolism
| Home | | Biochemistry |Chapter: Biochemistry : Glycogen Metabolism
The main stores of glycogen in the body are found in skeletal muscle, where they serve as a fuel reserve for the synthesis of ATP during muscle contraction, and in the liver, where they are used to maintain the blood glucose concentration, particularly during the early stages of a fast.
CHAPTER SUMMARY
The main stores of
glycogen in the body are found in skeletal muscle, where they serve as a fuel
reserve for the synthesis of ATP during muscle contraction, and in the liver,
where they are used to maintain the blood glucose concentration, particularly
during the early stages of a fast. Glycogen is a highly branched polymer of
α-D-glucose. The primary glycosidic bond is an α(1→4) linkage. After about
eight to ten glucosyl residues, there is a branch containing an α(1→6) linkage.
Uridine diphosphate (UDP)-glucose, the building block of glycogen, is
synthesized from glucose 1-phosphate and UTP by UDP-glucose pyrophosphorylase
(Figure 11.14). Glucose from UDP-glucose is transferred to the nonreducing ends
of glycogen chains by primer-requiring glycogen synthase, which makes α(1→4)
linkages. The primer is made by glycogenin. Branches are formed by
amylo-α(1→4)→α(1→6)-transglucosidase (common name, glucosyl 4:6 transferase),
which transfers a set of six to eight glucosyl residues from the nonreducing
end of the glycogen chain (breaking an α(1→4) linkage), and attaches it with an
α(1→6) linkage to another residue in the chain. Pyridoxal phosphate–requiring
glycogen phosphorylase cleaves the α(1→4) bonds between glucosyl residues at
the nonreducing ends of the glycogen chains, producing glucose 1-phosphate.
This sequential degradation continues until four glucosyl units remain before a
branch point. The resulting structure is called a limit dextrin that is
degraded by the bifunctional debranching enzyme.
Oligo-α(1→4)→α(1→4)-glucantransferase (common name, glucosyl 4:4 transferase)
removes the outer three of the four glucosyl residues at a branch and transfers
them to the nonreducing end of another chain, where they can be converted to
glucose 1-phosphate by glycogen phosphorylase. The remaining single glucose
residue attached in an α(1→6) linkage is removed hydrolytically by the amylo-(1→6)
glucosidase activity of debranching enzyme, releasing free glucose. Glucose
1-phosphate is converted to glucose 6-phosphate by phosphoglucomutase. In the
muscle, glucose 6-phosphate enters glycolysis. In the liver, the phosphate is
removed by glucose 6-phosphatase, releasing free glucose that can be used to
maintain blood glucose levels at the beginning of a fast. A deficiency of the
phosphatase causes glycogen storage disease Type 1a (Von Gierke disease). This
disease results in an inability of the liver to provide free glucose to the
body during a fast. It affects both glycogen degradation and gluconeogenesis.
Glycogen synthesis and degradation are reciprocally regulated to meet
whole-body needs by the same hormonal signals (namely, an elevated insulin
level results in overall increased glycogenesis and decreased glycogenolysis,
whereas an elevated glucagon, or epinephrine, level causes increased
glycogenolysis and decreased glycogenesis). Key enzymes are phosphorylated by a
family of protein kinases, some of which are cyclic adenosine monophosphate
dependent (a compound increased by glucagon and epinephrine). Phosphate groups
are removed by protein phosphatase-1 (active when its inhibitor is inactive in
response to elevated insulin levels). Glycogen synthase, phosphorylase kinase,
and phosphorylase are also allosterically regulated to meet tissues needs. In
the well-fed state, glycogen synthase is activated by glucose 6-phosphate, but
glycogen phosphorylase is inhibited by glucose 6-phosphate as well as by ATP.
In the liver, glucose also serves an an allosteric inhibitor of glycogen
phosphorylase. The Ca2+ released from the endoplasmic reticulum in
muscle during exercise and in liver in response to epinephrine activates
phosphorylase kinase by binding to the enzyme’s calmodulin subunit. This allows
the enzyme to activate glycogen phosphorylase, thereby causing glycogen
degradation. AMP activates glycogen phosphorylase in muscle.
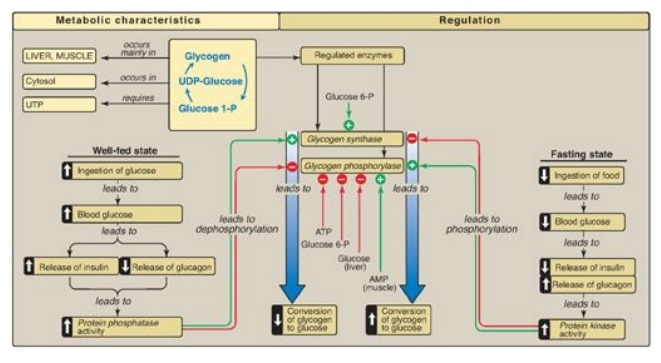
Figure 11.14 Key concept map
for glycogen metabolism in the liver. [Note: Glycogen phosphorylase is
phosphorylated by phosphorylase kinase, the “b” form of which can be activated
by calcium.] UDP = uridine diphosphate; UTP = uridine triphosphate; P = phosphate.
Study Questions
Choose the ONE best answer.
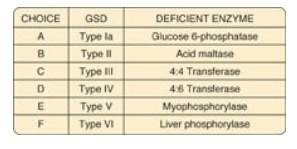
For Questions
11.1–11.4, match the deficient enzyme to the clinical finding in selected
glycogen storage diseases (GSDs).
11.1 Exercise intolerance, with no rise in blood
lactate during exercise
Correct answer = E. Myophosphorylase deficiency
prevents glycogen degradation in muscle, depriving muscle of glycogen-derived
glucose, resulting in decreased glycolysis and its anaerobic product, lactate.
Correct Answer = D. 4:6 Transferase (branching enzyme)
deficiency, a defect in glycogen synthesis, results in formation of glycogen
with fewer branches and decreased solubility.
Correct answer = B. Acid maltase [a(1→4)-glucosidase]
deficiency prevents degradation of any glycogen brought into lysosomes. A
variety of tissues are affected, with the most severe pathology resulting from
heart damage.
Correct answer = A. Glucose 6-phosphatase deficiency
prevents the liver from releasing free glucose into the blood, causing severe
fasting hypoglycemia, lacticacidemia, hyperuricemia, and hyperlipidemia.
11.2 Fatal, progressive
cirrhosis and glycogen with longer-than-normal outer chains
11.3 Generalized
accumulation of glycogen, severe hypotonia, and death from heart failure
11.4 Severe fasting
hypoglycemia, lacticacidemia, hyperuricemia, and hyperlipidemia
11.5 Epinephrine and glucagon have which one of the
following effects on hepatic glycogen metabolism?
A. Both glycogen
phosphorylase and glycogen synthase are activated by phosphorylation but at
significantly different rates.
B. Glycogen
phosphorylase is inactivated by the resuting rise in calcium, whereas glycogen
synthase is activated.
C. Glycogen phosphorylase is phosphorylated and
active, whereas glycogen synthase is phosphorylated and inactive.
D. The net synthesis of
glycogen is increased.
Correct answer = C. Epinephrine and glucagon both
cause increased glycogen degradation and decreased synthesis in the liver
through covalent modification (phosphorylation) of key enzymes of glycogen
metabolism. Glycogen phosphorylase is phosphorylated and active (“a” form),
whereas glycogen synthase is phosphorylated and inactive (“b” form). Glucagon
does not cause a rise in calcium.
11.6 In contracting skeletal muscle, a sudden
elevation of the sarcoplasmic calcium concentration will result in:
A. activation of cyclic
adenosine monophosphate (cAMP)-dependent protein kinase A.
B. conversion of cAMP
to AMP by phosphodiesterase.
C. direct activation of
glycogen synthase b.
D. direct activation of phosphorylase kinase b.
E. inactivation of
phosphorylase kinase a by the action of protein phosphatase-1.
Correct answer = D. Ca2+ released from the
sarcoplasmic reticulum during exercise binds to the calmodulin subunit of
phosphorylase kinase, thereby allosterically activating the “b” form of this
enzyme. The other choices are not caused by an elevation of cytosolic calcium.
11.7 Explain why the hypoglycemia seen with Type Ia
glycogen storage disease (glucose 6-phosphatase deficiency) is severe, whereas
that seen with Type VI (liver phosphorylase deficiency) is mild.
With Type Ia, the liver
is unable to generate free glucose either from glycogenolysis or
gluconeogenesis because both processes produce glucose 6-phosphate. With Type
VI, the liver is still able to produce free glucose from gluconeogenesis, but
glycogenolysis is inhibited.
Related Topics
