Galactose Metabolism
| Home | | Biochemistry |Chapter: Biochemistry : Metabolism of Monosaccharides and Disaccharides
The major dietary source of galactose is lactose (galactosyl β-1,4-glucose) obtained from milk and milk products.
GALACTOSE METABOLISM
The major dietary
source of galactose is lactose (galactosyl β-1,4-glucose) obtained from milk
and milk products. [Note: The digestion of lactose by β-galactosidase (lactase)
of the intestinal mucosal cell membrane.] Some galactose can also be obtained
by lysosomal degradation of complex carbohydrates, such as glycoproteins and
glycolipids, which are important membrane components. Like fructose, the
transport of galactose into cells is not insulin dependent.
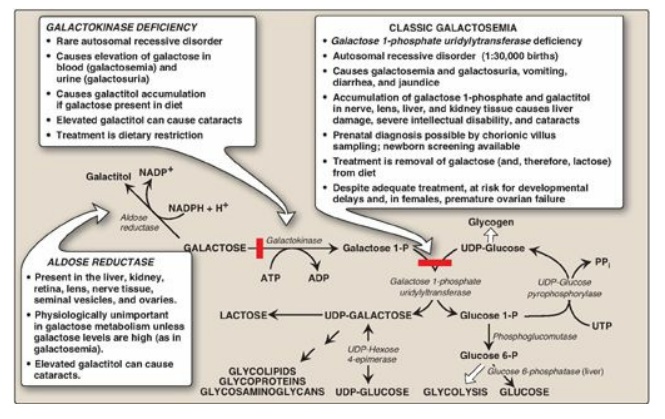
Figure 12.5 Metabolism of galactose. UDP = uridine diphosphate; UTP = uridine triphosphate; P = phosphate; PPi = pyrophosphate; NADP(H) = nicotinamide adenine dinucleotide phosphate; ADP = adenosine diphosphate.
A. Phosphorylation of galactose
Like fructose, galactose must be phosphorylated before it can be further metabolized. Most tissues have a specific enzyme for this purpose, galactokinase, which produces galactose 1-phosphate (Figure 12.5). As with other kinases, ATP is the phosphate donor.
B. Formation of uridine diphosphate-galactose
Galactose 1-phosphate
cannot enter the glycolytic pathway unless it is first converted to uridine
diphosphate (UDP)-galactose (Figure 12.6). This occurs in an exchange reaction,
in which UDP-glucose reacts with galactose 1-phosphate, producing UDP-galactose
and glucose 1-phosphate (see Figure 12.5). The enzyme that catalyzes this reaction
is galactose 1-phosphate uridylyltransferase (GALT).
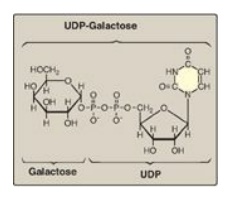
Figure 12.6 Structure of UDP-galactose. UDP = uridine diphosphate.
C. Use of uridine diphosphate-galactose as a carbon source for glycolysis or gluconeogenesis
For UDP-galactose to enter the mainstream of glucose metabolism, it must first be converted to its C-4 epimer, UDP-glucose, by UDP-hexose 4-epimerase. This “new” UDP-glucose (produced from the original UDP-galactose) can then participate in many biosynthetic reactions as well as being used in the GALT reaction described above. (See Figure 12.5 for a summary of this interconversion.)
D. Role of uridine diphosphate-galactose in biosynthetic reactions
UDP-galactose can serve
as the donor of galactose units in a number of synthetic pathways, including
synthesis of lactose (see below), glycoproteins, glycolipids, and
glycosaminoglycans. [Note: If galactose is not provided by the diet (for
example, when it cannot be released from lactose as a result of a lack of
β-galactosidase in people who are lactose intolerant), all tissue requirements
for UDP-galactose can be met by the action of UDP-hexose 4-epimerase on
UDP-glucose, which is efficiently produced from glucose 1-phosphate (see Figure
12.5).]
E. Disorders of galactose metabolism
GALT is deficient in
individuals with classic galactosemia (see Figure 12.5). In this disorder,
galactose 1-phosphate and, therefore, galactose accumulate. Physiologic
consequences are similar to those found in hereditary fructose intolerance, but
a broader spectrum of tissues is affected. The accumulated galactose is shunted
into side pathways such as that of galactitol production. This reaction is
catalyzed by aldose reductase, the same enzyme that converts glucose to
sorbitol. Treatment requires removal of galactose and lactose from the diet.
GALT deficiency is part of the newborn screening panel. [Note: A deficiency in
galactokinase results in a less severe disorder of galactosemia metabolism,
although cataracts are common (see Figure 12.5).]
Related Topics
