Glucose 6-Phosphate Dehydrogenase Deficiency
| Home | | Biochemistry |Chapter: Biochemistry : Pentose Phosphate Pathway and Nicotinamide Adenine Dinucleotide Phosphate
G6PD deficiency is a hereditary disease characterized by hemolytic anemia caused by the inability to detoxify oxidizing agents.
GLUCOSE 6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is a
hereditary disease characterized by hemolytic anemia caused by the inability to
detoxify oxidizing agents. G6PD deficiency is the most common disease-producing
enzyme abnormality in humans, affecting more than 400 million individuals
worldwide. This deficiency has the highest prevalence in the Middle East,
tropical Africa and Asia, and parts of the Mediterranean. G6PD deficiency is X
linked and is, in fact, a family of deficiencies caused by a number of
different mutations in the gene coding for G6PD. Only some of the resulting
protein variants cause clinical symptoms. [Note: In addition to hemolytic
anemia, a clinical manifestation of G6PD deficiency is neonatal jaundice
appearing 1–4 days after birth. The jaundice, which may be severe, typically
results from increased production of unconjugated bilirubin.] The life span of
individuals with a severe form of G6PD deficiency may be somewhat shortened as
a result of complications arising from chronic hemolysis. This negative effect
of G6PD deficiency has been balanced in evolution by an advantage in survival—an
increased resistance to Plasmodium falciparum malaria. [Note: Sickle cell trait
and β-thalassemia minor also confer resistance to malaria.]
A. Role of glucose 6-phosphate dehydrogenase in red blood cells
Diminished G6PD
activity impairs the ability of the cell to form the NADPH that is essential
for the maintenance of the G-SH pool. This results in a decrease in the
cellular detoxification of free radicals and peroxides formed within the cell
(Figure 13.10). G-SH also helps maintain the reduced states of sulfhydryl
groups in proteins, including hemoglobin. Oxidation of those sulfhydryl groups
leads to the formation of denatured proteins that form insoluble masses (called
Heinz bodies) that attach to RBC membranes (Figure 13.11). Additional oxidation
of membrane proteins causes RBCs to be rigid (less deformable), and they are
removed from the circulation by macrophages in the spleen and liver. Although
G6PD deficiency occurs in all cells of the affected individual, it is most
severe in RBCs, where the pentose phosphate pathway provides the only means of
generating NADPH. Other tissues have alternative sources for NADPH production
(such as NADP+-dependent malate dehydrogenase [malic enzyme];) that can keep
G-SH reduced. The RBC has no nucleus or ribosomes and cannot renew its supply
of the enzyme. Thus, RBCs are particularly vulnerable to enzyme variants with
diminished stability.
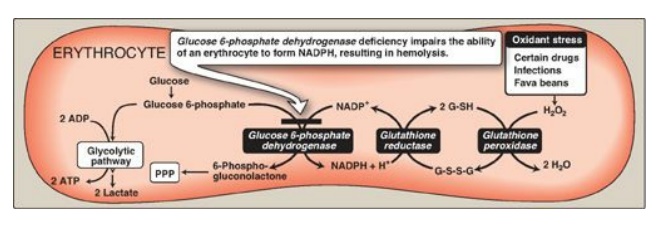
Figure 13.10 Pathways of glucose 6-phosphate metabolism in the erythrocyte. NADP(H) nicotinamide adenine dinucleotide phosphate; G-SH = reduced glutathionine; G-S-S-G oxidized glutathionine; PPP = pentose phosphate pathway.
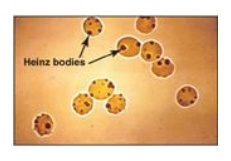
Figure 13.11 Heinz bodies in
erythrocytes of a patient with glucose 6-phosphate dehydrogenase deficiency.
B. Precipitating factors in glucose 6-phosphate dehydrogenase deficiency
Most individuals who
have inherited one of the G6PD mutations do not show clinical manifestations
(that is, they are asymptomatic). However, some patients with G6PD deficiency
develop hemolytic anemia if they are treated with an oxidant drug, ingest fava
beans, or contract a severe infection.
1. Oxidant drugs: Commonly used drugs that produce
hemolytic anemia in patients with G6PD deficiency are best remembered from the
mnemonic AAA: antibiotics (for example, sulfamethoxazole and chloramphenicol),
antimalarials (for example, primaquine but not chloroquine or quinine), and
antipyretics (for example, acetanilid but not acetaminophen).
2. Favism: Some forms of G6PD deficiency, for example the
Mediterranean variant, are particularly susceptible to the hemolytic effect of
the fava (broad) bean, a dietary staple in the Mediterranean region. Favism,
the hemolytic effect of ingesting fava beans, is not observed in all
individuals with G6PD deficiency, but all patients with favism have G6PD
deficiency.
3. Infection: Infection is the most common precipitating factor of hemolysis in G6PD deficiency. The inflammatory response to infection results in the generation of free radicals in macrophages, which can diffuse into the RBC and cause oxidative damage.
C. Properties of the variant enzymes
Almost all G6PD
variants are caused by point mutations in the gene for G6PD. Some mutations do
not disrupt the structure of the enzyme’s active site and, therefore, do not
affect enzymic activity. However, many mutant enzymes show altered kinetic
properties. For example, variant enzymes may show decreased catalytic activity,
decreased stability, or an alteration of binding affinity for NADP +, NADPH, or
glucose 6-phosphate. The severity of the disease usually correlates with the
amount of residual enzyme activity in the patient’s RBC. For example, variants
can be classified as shown in Figure 13.12. G6PD A- is the prototype
of the moderate (Class III) form of the disease. The RBCs contain an unstable
but kinetically normal G6PD, with most of the enzyme activity present in the
reticulocytes and younger RBCs (Figure 13.13). The oldest RBCs, therefore, have
the lowest level of enzyme activity and are preferentially removed in a
hemolytic episode. G6PD Mediterranean is the prototype of a more severe (Class
II) deficiency in which the enzyme has decreased stability resulting in
decreased enzymic activity. Class I mutations (rare) are the most severe and
are associated with chronic nonspherocytic hemolytic anemia, which occurs even
in the absence of oxidative stress.
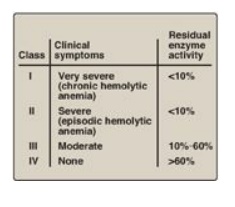
Figure 13.12 Classification of glucose 6-phosphate dehydrogenase (G6PD) deficiency variants. Note: Class V variants (not shown in table) result in overproduction of G6PD.
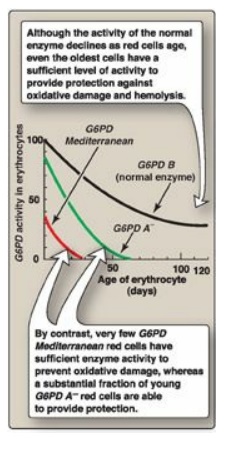
Figure 13.13 Decline of
erythrocyte glucose 6-phosphate dehydrogenase (G6PD) activity with cell age for
the three most commonly encountered forms of the enzyme.
D. Molecular biology of glucose 6-phosphate dehydrogenase
The cloning of the gene
for G6PD and the sequencing of its DNA have permitted the identification of
mutations that cause G6PD deficiency. More than 400 different G6PD variants
have been identified, a finding that explains the numerous biochemical and
clinical phenotypes that have been described. Most mutations that result in
enzymic deficiency are missense mutations in the coding region. Both G6PD A-
and G6PD Mediterranean represent mutant enzymes that differ from the respective
normal variants by a single amino acid. Large deletions or frameshift mutations
have not been identified, suggesting that complete absence of G6PD activity is
probably lethal.
Related Topics
