Degradation of Glycosaminoglycans
| Home | | Biochemistry |Chapter: Biochemistry : Glycosaminoglycans, Proteoglycans, and Glycoproteins
GAGs are degraded in lysosomes, which contain hydrolytic enzymes that are most active at a pH of approximately 5. Therefore, as a group, these enzymes are called acid hydrolases.
DEGRADATION OF GLYCOSAMINOGLYCANS
GAGs are degraded in
lysosomes, which contain hydrolytic enzymes that are most active at a pH of
approximately 5. Therefore, as a group, these enzymes are called acid
hydrolases. [Note: The low pH optimum is a protective mechanism that prevents
the enzymes from destroying the cell should leakage occur into the cytosol
where the pH is neutral.] The half-lives of GAGs vary from minutes to months
and are influenced by the type of GAG and its location in the body.
A. Phagocytosis of extracellular glycosaminoglycans
Because GAGs are
extracellular or cell-surface compounds, they must first be engulfed by an
invagination of the cell membrane (phagocytosis), forming a vesicle inside of
which the GAGs are to be degraded. This vesicle then fuses with a lysosome,
forming a single digestive vesicle in which the GAGs are efficiently degraded
(see: for a discussion of phagocytosis).
B. Lysosomal degradation of glycosaminoglycans
The lysosomal
degradation of GAGs requires a large number of acid hydrolases for complete
digestion. First, the polysaccharide chains are cleaved by endoglycosidases,
producing oligosaccharides. Further degradation of the oligosaccharides occurs
sequentially from the nonreducing end of each chain, the last group (sulfate or
sugar) added during synthesis being the first group removed (by sulfatases or
exoglycosidases). Examples of some of these enzymes and the bonds they
hydrolyze are shown in Figure 14.12. [Note: Endo- and exoglycosidases are also
involved in the lysosomal degradation of glycoproteins and glycolipids.
Deficiencies in these enzymes result in the accumulation of partially degraded
carbohydrates, resulting in tissue damage.]
Multiple sulfatase deficiency is a rare lysosomal
storage disease in which all sulfatases are nonfunctional due to a defect in
the formation of formylglycine, an amino acid derivative required at the active
site for enzymic activity to occur.
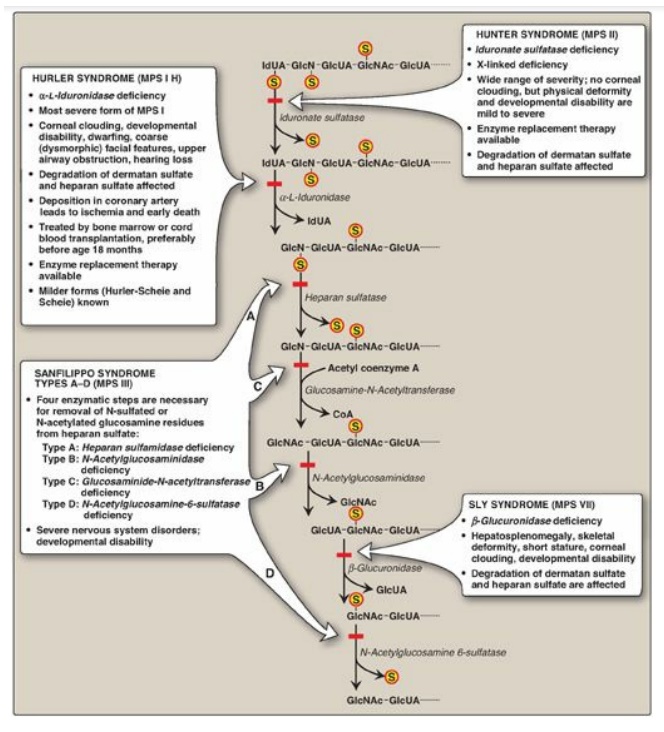
Figure 14.12 Degradation of the glycosaminoglycan heparan sulfate by lysosomal enzymes, indicating sites of enzyme deficiencies in some representative mucopolysaccharidoses (MPSs). [Note: Deficiencies in the degradation of keratan sulfate result in Morquio syndrome, A and B. Deficiencies in the degradation of dermatan sulfate result in Maroteaux-Lamy syndrome.] GlcUA = glucuronic acid; IdUA = iduronic acid; GalNAc = N-acetylgalactosamine; GlcNAc = N-acetylglucosamine; GlcN = glucosamine; S = sulfate.
Related Topics
