Chapter Summary, Questions Answers - Glycosaminoglycans, Proteoglycans, and Glycoproteins
| Home | | Biochemistry |Chapter: Biochemistry : Glycosaminoglycans, Proteoglycans, and Glycoproteins
Glycosaminoglycans (GAGs) are long, negatively charged, unbranched, heteropolysaccharide chains generally composed of a repeating disaccharide unit [acidic sugar–amino sugar]n.
CHAPTER SUMMARY
Glycosaminoglycans
(GAGs) are long, negatively charged, unbranched, heteropolysaccharide chains
generally composed of a repeating disaccharide unit [acidic sugar–amino sugar]n
(Figure 14.18). The amino sugar is either D glucosamine or D-galactosamine in
which the amino group is usually acetylated, thus eliminating its positive
charge. The amino sugar may also be sulfated on carbon 4 or 6 or on a
nonacetylated nitrogen. The acidic sugar is either D-glucuronic acid or its C-5
epimer L-iduronic acid. GAGs bind large amounts of water, thereby producing the
gel-like matrix that forms the basis of the body’s ground substance. The
viscous, lubricating properties of mucous secretions are also caused by the
presence of GAGs, which led to the original naming of these compounds as
mucopolysaccharides. There are six major types of GAGs, including chondroitin
4- and 6-sulfates, keratan sulfate, dermatan sulfate, heparin, heparan sulfate,
and hyaluronic acid. All of the GAGs, except hyaluronic acid, are found
covalently attached to protein, forming proteoglycan monomers, which consist of
a core protein to which the linear GAG chains are covalently attached. The
proteoglycan monomers associate with a molecule of hyaluronic acid to form
proteoglycan aggregates. GAGs are synthesized in the Golgi. The polysaccharide
chains are elongated by the sequential addition of alternating acidic and amino
sugars, donated by their UDP-derivatives. D-glucuronate may be epimerized to
L-iduronate. The last step in synthesis is sulfation of some of the amino
sugars. The source of the sulfate is 3′-phosphoadenosyl-5′-phosphosulfate. The
completed proteoglycans are secreted into the extracellular matrix or remain
associated with the outer surface of cells. GAGs are degraded by lysosomal acid
hydrolases. They are first broken down to oligosaccharides, which are degraded
sequentially from the nonreducing end of each chain. A deficiency of any one of
the hydrolases results in a mucopolysaccharidosis. These are hereditary
disorders in which GAGs accumulate in tissues, causing symptoms such as
skeletal and extracellular matrix deformities and intellectual disability.
Examples of these genetic diseases include Hunter and Hurler syndromes.
Glycoproteins are proteins to which oligosaccharides are covalently attached.
They differ from the proteoglycans in that the length of the glycoprotein’s
carbohydrate chain is relatively short (usually two to ten sugar residues long,
although they can be longer), may be branched, and does not contain serial
disaccharide units. Membrane-bound glycoproteins participate in a broad range
of cellular phenomena, including cell-surface recognition (by other cells,
hormones, and viruses), cell-surface antigenicity (such as the blood group
antigens), and as components of the extracellular matrix and of the mucins of
the gastrointestinal and urogenital tracts, where they act as protective
biologic lubricants. In addition, almost all of the globular proteins present
in human plasma are glycoproteins. Glycoproteins are synthesized in the rough
endoplasmic reticulum (RER) and t h e Golgi. The precursors of the carbohydrate
components of glycoproteins are nucleotide sugars. O-linked glycoproteins are
synthesized in the Golgi by the sequential transfer of sugars from their
nucleotide carriers to the hydroxyl group of a serine or threonine residue in
the protein. N-linked glycoproteins contain varying amounts of mannose. They
are synthesized by the transfer of a preformed oligosaccharide from its ER
membrane lipid carrier, dolichol pyrophosphate, to the amide N of an asparagine
residue in the protein. A deficiency in the phosphorylation of mannose residues
in N-linked glycoprotein enzymes destined for the lysosomes results in I-cell
disease. Glycoproteins are degraded in lysosomes by acid hydrolases. A
deficiency of any one of these enzymes results in a lysosomal glycoprotein
storage disease (oligosaccharidosis), resulting in accumulation of partially
degraded structures in the lysosome.
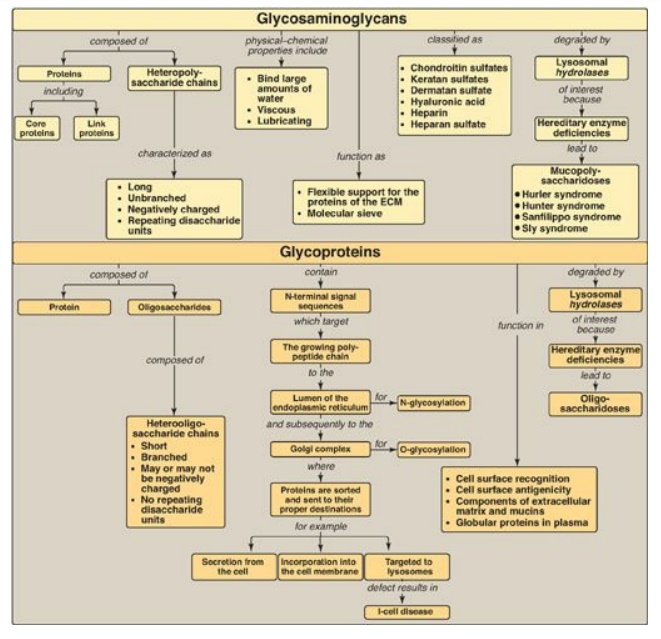
Figure 14.18 Key concept map
for glycosaminoglycans and glycoproteins. ECM = extracellular matrix.
Study Questions
Choose the ONE best answer.
14.1 Mucopolysaccharidoses are hereditary lysosomal
storage diseases. They are caused by:
A. defects in the degradation of
glycosaminoglycans.
B. defects in the
targeting of enzymes to lysosomes.
C. an increased rate of
synthesis of the carbohydrate component of proteoglycans.
D. an insufficient rate
of synthesis of proteolytic enzymes.
E. the synthesis of
abnormally small amounts of core proteins.
F. the synthesis of
heteropolysaccharides with an altered structure.
Correct answer = A. The mucopolysaccharidoses are
caused by deficiencies in any one of the lysosomal acid hydrolases responsible
for the degradation of glycosaminoglycans (not proteins). The enzyme is
correctly targeted to the lysosome, so blood levels of the enzyme do not
increase, but it is nonfunctional. In these diseases, synthesis of the protein
and carbohydrate components of proteoglycans is unaffected, both in terms of
structure and amount.
14.2 The presence of the following compound in the
urine of a patient suggests a deficiency in which one of the enzymes listed
below?

A. Galactosidase
B. Glucuronidase
C. Iduronidase
D. Mannosidase
E. Sulfatase
Correct answer = E. Degradation of glycoproteins
follows the rule “last on, first off.” Because sulfation is the last step in
the synthesis of this sequence, a sulfatase is required for the next step in
the degradation of the compound shown.
14.3 An 8-month-old boy with coarse facial
features, skeletal abnormalities, and delays in both growth and development is
diagnosed with I-cell disease based on his presentation and on histologic and
biochemical testing. I-cell disease is characterized by:
A. decreased production
of cell-surface O-linked glycoproteins.
B. elevated levels of acid hydrolases in the blood.
C. an inability to
N-glycosylate proteins.
D. increased synthesis
of proteoglycans.
E. oligosaccharides in
the urine.
Correct answer = B. I-cell disease is a lysosomal storage
disease caused by deficiency of a protein essential for synthesis of the
mannose 6-phosphate signal that targets acid hydrolases to the lysosome. This
results in secretion of these enzymes from the cell and accumulation of
materials within the lysosome due to impaired degradation. None of the other
choices relate in any way to I-cell disease or lysosomal function.
Oligosaccharides in the urine are characteristic of the muco-and
polysaccharidoses but not I-cell disease (a mucolipidosis).
14.4 An infant with corneal clouding has dermatan
sulfate and heparin sulfate in his urine. Decreased activity of which of the
enzymes listed below would confirm the suspected diagnosis of Hurler syndrome?
A. α-L-Iduronidase
B. β-Glucuronidase
C. Glycosyltransferase
D. Iduronate sulfatase
Correct answer = A. Hurler syndrome, a defect in the
lysosomal degradation of glycosaminoglycans (GAGs) with corneal clouding, is
due to a deficiency in α-L-iduronidase. β-glucuronidase is deficient in Sly
syndrome, and iduronate sulfatase is deficient in Hunter syndrome.
Glycosyltransferases are enzymes of GAG synthesis.
14.5 Distinguish between glycoproteins and
proteoglycans.
Glycoproteins are
proteins to which short, branched, oligosaccharide chains are attached.
Proteoglycans consist of a core protein to which long, unbranched,
glycosaminoglycan (GAG) chains are attached. GAGs are large complexes of
negatively charged heteropolysaccharides composed of repeating [acidic sugar–amino
sugar]n disaccharide units.
Related Topics
