Mobilization of Stored Fats and Oxidation of Fatty Acids
| Home | | Biochemistry |Chapter: Biochemistry : Fatty Acid, Ketone Body, and Triacylglycerol Metabolism
Fatty acids stored in WAT, in the form of neutral TAG, serve as the body’s major fuel storage reserve. TAGs provide concentrated stores of metabolic energy because they are highly reduced and largely anhydrous.
MOBILIZATION OF STORED FATS AND OXIDATION OF FATTY
ACIDS
Fatty acids stored in
WAT, in the form of neutral TAG, serve as the body’s major fuel storage
reserve. TAGs provide concentrated stores of metabolic energy because they are
highly reduced and largely anhydrous. The yield from the complete oxidation of
fatty acids to CO2 and H2O is 9 kcal/g fat (as compared
to 4 kcal/g protein or carbohydrate, see Figure 27.5).
A. Release of fatty acids from fat
The mobilization of
stored fat requires the hydrolytic release of fatty acids and glycerol from
their TAG form. This process of lipolysis is achieved by lipases. It is
initiated by adipose triglyceride lipase (ATGL), which generates a
diacylglycerol that is the preferred substrate for hormone-sensitive lipase
(HSL). The monoacylglycerol (MAG) product of HSL is acted upon by MAG lipase.
1. Regulation of hormone-sensitive lipase: HSL is active when phosphorylated
by PKA, a 3′,5′-cyclic AMP(cAMP)–dependent protein kinase. cAMP is produced in
the adipocyte when catecholamines (such as epinephrine) bind to cell membrane
β-adrenergic receptors and activate adenylyl cyclase (Figure 16.15). The
process is similar to that of the activation of glycogen phosphorylase (see
Figure 11.9). [Note: Because ACC is inhibited by hormone-directed
phosphorylation, when the cAMP-mediated cascade is activated (see Figure 16.8),
fatty acid synthesis is turned off and TAG degradation is turned on.] In the
presence of high plasma levels of insulin, HSL is dephosphorylated and
inactivated. Insulin also suppresses expression of ATGL. [Note: Fat droplets
are coated by a protein (perilipin) that limits access of HSL. Phosphorylation
of perilipin by PKA allows translocation and binding of HSL to the droplet.]
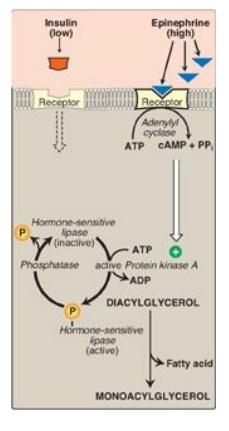
Figure 16.15 Hormonal regulation of fat degradation in the adipocyte. [Note: Triacylglycerol is degraded to diacylglycerol by adipose triglyceride lipase.] cAMP = cyclic adenosine monophosphate; PPi = pyrophosphate; ADP = adenosine diphosphate; = phosphate.
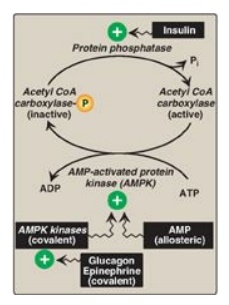
Figure 16.8 Covalent regulation (phosphorylation) of acetyl CoA carboxylase by AMPK, which itself is regulated both covalently and allosterically. CoA = coenzyme A; ADP = adenosine diphosphate; P = phosphate; Pi = inorganic phosphate; AMP = adenosine monophosphate.
2. Fate of glycerol: The glycerol released during TAG
degradation cannot be metabolized by adipocytes because they lack glycerol
kinase. Rather, glycerol is transported through the blood to the liver, where
it can be phosphorylated. The resulting glycerol 3-phosphate can be used to
form TAG in the liver or can be converted to DHAP by reversal of the glycerol
3-phosphate dehydrogenase reaction illustrated in Figure 16.13. DHAP can
participate in glycolysis or gluconeogenesis.
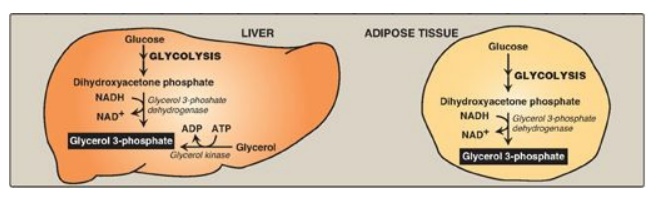
Figure 16.13 Pathways for production of glycerol 3-phosphate in liver and adipose tissue. [Note: Glycerol 3-phosphate can also be generated by glyceroneogenesis.] NAD(H) = nicotinamide adenine dinucleotide; ADP = adenosine diphosphate
3. Fate of fatty acids: The free (unesterified) fatty acids move through the cell membrane of the adipocyte and bind to plasma albumin. They are transported to the tissues, enter cells, get activated to their CoA derivatives, and are oxidized for energy in mitochondria. Regardless of their levels, plasma FFAs cannot be used for fuel by red blood cells (RBCs), which have no mitochondria. Brain, too, does not use fatty acids for energy, but the reasons are less clear. [Note: Over 50% of the fatty acids released from adipose TAG are reesterified to glycerol 3-phosphate. WAT does not express glycerol kinase, and the phosphorylated glycerol is produced by glyceroneogenesis, an incomplete version of gluconeogenesis: pyruvate to OAA via pyruvate carboxylase and OAA to phosphoenolpyruvate (PEP) via phosphoenolpyruvate carboxykinase. The PEP is converted (by reactions common to glycolysis and gluconeogenesis) to DHAP, which is reduced to glycerol 3-phosphate. The process reduces plasma FFAs, molecules associated with insulin resistance in type 2 diabetes and obesity.]
B. β-Oxidation of fatty acids
The major pathway for
catabolism of fatty acids is a mitochondrial pathway called β-oxidation, in
which two-carbon fragments are successively removed from the carboxyl end of
the fatty acyl CoA, producing acetyl CoA, NADH, and flavin adenine dinucleotide
(FADH2).
1. Transport of long-chain fatty acids into
mitochondria: After
a LCFA enters a cell, it is converted in the cytosol to its CoA derivative by
long-chain fatty acyl CoA synthetase (thiokinase), an enzyme of the outer mitochondrial
membrane. Because β-oxidation occurs in the mitochondrial matrix, the fatty
acid must be transported across the inner mitochondrial membrane that is
impermeable to CoA. Therefore, a specialized carrier transports the long-chain
acyl group from the cytosol into the mitochondrial matrix. This carrier is
carnitine, and this rate-limiting transport process is called the “carnitine
shuttle” (Figure 16.16).
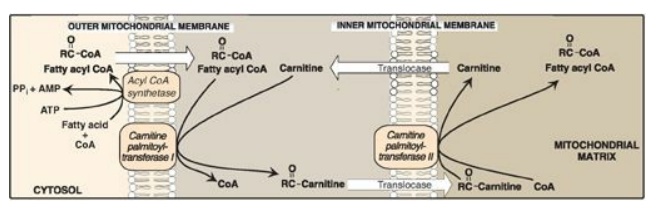
Figure 16.16 Carnitine
shuttle. The net effect is that a long-chain fatty acyl coenzyme A (CoA) is transported from the
outside to the inside of mitochondria. AMP = adenosine monophosphate; PPi =
pyrophosphate.
a. Steps in translocation: First, the acyl group is
transferred from CoA to carnitine by carnitine palmitoyltransferase I (CPT-I),
an enzyme of the outer mitochondrial membrane. [Note: CPT-I is also known as
CAT-I for carnitine acyltransferase I.] This reaction forms an acylcarnitine
and regenerates free CoA. Second, the acylcarnitine is transported into the
mitochondrial matrix in exchange for free carnitine by carnitine–acylcarnitine
translocase. Carnitine palmitoyltransferase II (CPT-II, or CAT-II), an enzyme
of the inner mitochondrial membrane, catalyzes the transfer of the acyl group
from carnitine to CoA in the mitochondrial matrix, thus regenerating free
carnitine.
b. Inhibitor of the carnitine shuttle: Malonyl CoA inhibits CPT-I, thus
preventing the entry of long-chain acyl groups into the mitochondrial matrix.
Therefore, when fatty acid synthesis is occurring in the cytosol (as indicated
by the presence of malonyl CoA), the newly made palmitate cannot be transferred
into mitochondria and degraded. [Note: Muscle, although it does not synthesize
fatty acids, contains the mitochondrial isoform of ACC (ACC2 ), allowing muscle
to regulate β-oxidation.] Fatty acid oxidation is also regulated by the acetyl
CoA to CoA ratio: as the ratio increases, the CoA-requiring thiolase reaction
decreases (Figure 16.17).
c. Sources of carnitine: Carnitine can be obtained from the
diet, where it is found primarily in meat products. Carnitine can also be
synthesized from the amino acids lysine and methionine by an enzymatic pathway
found in the liver and kidney but not in skeletal or heart muscle. Therefore,
these latter tissues are totally dependent on uptake of carnitine provided by
endogenous synthesis or the diet and distributed by the blood. [Note: Skeletal
muscle contains about 97% of all carnitine in the body.]
d. Carnitine deficiencies: Such deficiencies result in a
decreased ability of tissues to use LCFAs as a fuel. Primary carnitine
deficiency is caused by defects in a membrane transporter that prevent uptake
of carnitine by cardiac and skeletal muscle and kidney. Treatment includes
carnitine supplementation. Secondary carnitine deficiency occurs primarily as a
result of defects in fatty acid oxidation leading to the accumulation of
acylcarnitines that are excreted in the urine, decreasing carnitine
availability. Acquired secondary carnitine deficiency can be seen, for example,
in patients with liver disease (decreased carnitine synthesis) or those taking
the antiseizure drug valproic acid (decreased renal reabsorption). [Note:
Defects in mitochondrial oxidation can also be caused by deficiencies in CPT-I
and CPT-II. CPT-I deficiency affects the liver, where an inability to use LCFAs
for fuel greatly impairs that tissue’s ability to synthesize glucose (an
endergonic process) during a fast. This can lead to severe hypoglycemia, coma,
and death. CPT-II deficiency can affect the liver and cardiac and skeletal
muscle. The most common (and least severe) form affects skeletal muscle. It
presents as muscle weakness with myoglobinemia following prolonged exercise.
Treatment includes avoidance of fasting and adopting a diet high in carbohydrates
and low in fat but supplemented with medium-chain TAGs.]
2. Entry of short- and medium-chain fatty acids
into the mitochondria:
Fatty acids shorter than 12 carbons can cross the inner mitochondrial membrane
without the aid of carnitine or the CPT system. Once inside the mitochondria,
they are activated to their CoA derivatives by matrix enzymes, and are
oxidized. [Note: Medium-chain fatty acids are plentiful in human milk. Because
their oxidation is not dependent on CPT-I, it is not subject to inhibition by
malonyl CoA.]
3. Reactions of β-oxidation: The first cycle of β-oxidation is
shown in Figure 16.17. It consists of a sequence of four reactions involving
the β-carbon (carbon 3) that results in shortening the fatty acid chain by two
carbons at the carboxylate end. The steps include an oxidation that produces FADH2,
a hydration step, a second oxidation that produces NADH, and a thiolytic
cleavage that releases a molecule of acetyl CoA. Each step is catalyzed by
enzymes with chain-length specificity. These four steps are repeated for
saturated fatty acids of even-numbered carbon chains (n/2) - 1 times (where n
is the number of carbons), each cycle producing one acetyl CoA plus one NADH
and one FADH2. The acetyl CoA can be oxidized or used in hepatic
ketogenesis (see below). The reduced coenzymes are oxidized by the electron
transport chain. The final thiolytic cleavage produces two acetyl groups.
[Note: Acetyl CoA is a positive allosteric effector of pyruvate carboxylase,
thus linking fatty acid oxidation and gluconeogenesis.]
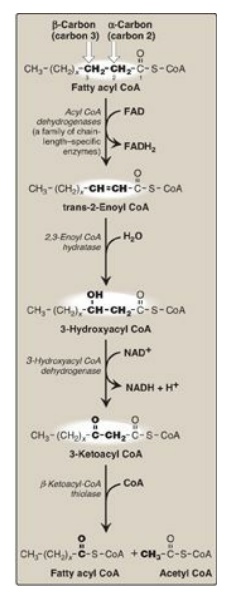
Figure 16.17 Enzymes involved
in the β-oxidation of fatty acyl coenzyme A (CoA). [Note: 2,3-Enoyl CoA
hydratase requires a trans double bond between carbon 2 and carbon 3.] FAD(H2)
= flavin adenine dinucleotide; NAD(H) = nicotinamide adenine dinucleotide.
4. Energy yield from fatty acid oxidation: The energy yield from the
β-oxidation pathway is high. For example, the oxidation of a molecule of
palmitoyl CoA to CO2 and H2O produces 8 acetyl CoA, 7
NADH, and 7 FADH2, from which 131 ATP can be generated. However,
activation of the fatty acid requires 2 ATP. Therefore, the net yield from
palmitate is 129 ATP (Figure 16.18). A comparison of the processes of synthesis
and degradation of long-chain saturated fatty acids with an even number of
carbon atoms is provided in Figure 16.19.
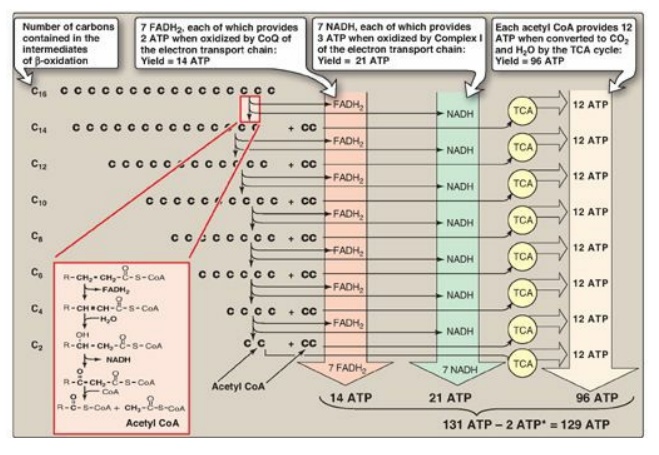
Figure 16.18 Summary of the energy yield from the oxidation of palmitoyl coenzyme A (CoA) (16 carbons). *Activation of palmitate to palmitoyl CoA requires the equivalent of 2 ATP (ATP → AMP + PPi). FADH2 = flavin adenine dinucleotide; NADH = nicotinamide adenine dinucleotide; TCA = tricarboxylic acid; CoQ = coenzyme Q.
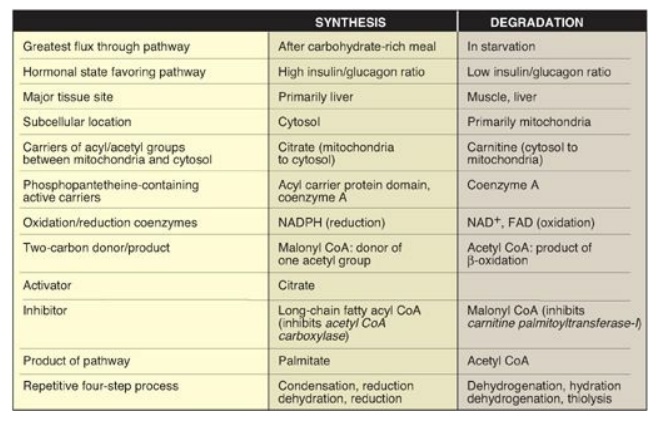
Figure 16.19 Comparison of the synthesis and degradation of long-chain, even-numbered, saturated fatty acids. NADPH = nicotinamide adenine dinucleotide phosphate; NAD = nicotinamide adenine dinucleotide; FAD = flavin adenine dinucleotide; CoA = coenzyme A.
5. Medium-chain fatty acyl CoA dehydrogenase deficiency: In mitochondria, there are four fatty acyl CoA dehydrogenase species, each with distinct but overlapping specificity for either short-, medium-, long-, or VLCFAs. Medium-chain fatty acyl CoA dehydrogenase (MCAD) deficiency, an autosomal-recessive disorder, is one of the most common inborn errors of metabolism and the most common inborn error of fatty acid oxidation, being found in 1:14,000 births worldwide, with a higher incidence in Caucasians of Northern European descent. It results in decreased ability to oxidize fatty acids with six to ten carbons (which accumulate and can be measured in urine), severe hypoglycemia (because the tissues must increase their reliance on glucose), and hypoketonemia (because of decreased production of acetyl CoA). See below. Treatment includes avoidance of fasting. MCAD deficiency has been identified as the cause of some cases originally reported as sudden infant death syndrome or Reye syndrome.
6. Oxidation of fatty acids with an odd number of
carbons: This
process proceeds by the same reaction steps as that of fatty acids with an even
number of carbons, until the final three carbons are reached. This compound,
propionyl CoA, is metabolized by a three-step pathway (Figure 16.20). [Note:
Propionyl CoA is also produced during the metabolism of certain amino acids
(see Figure 20.10).]
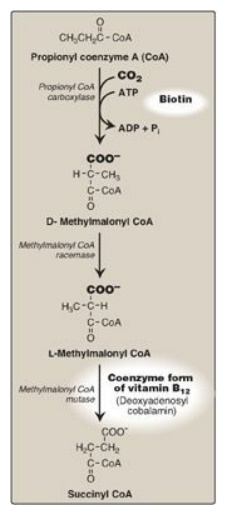
Figure 16.20 Metabolism of propionyl CoA. ADP = adenosine diphosphate; Pi = inorganic phosphate.
a. Synthesis of D-methylmalonyl coenzyme A: First, propionyl CoA is carboxylated, forming D-methylmalonyl coenzyme A. The enzyme propionyl CoA carboxylase has an absolute requirement for the coenzyme biotin, as do ACC and most other carboxylases.
b. Formation of L-methylmalonyl coenzyme A: Next, the D-isomer is converted to
the L-form by the enzyme, methylmalonyl CoA racemase.
c. Synthesis of succinyl coenzyme A: Finally, the carbons of
L-methylmalonyl CoA are rearranged, forming succinyl CoA, which can enter the
tricarboxylic acid (TCA) cycle. [Note: This is the only example of a glucogenic
precursor generated from fatty acid oxidation.] The enzyme methylmalonyl CoA
mutase requires a coenzyme form of vitamin B12
(deoxyadenosylcobalamin). The mutase reaction is one of only two reactions in
the body that require vitamin B12. [Note: In patients with vitamin B12
deficiency, both propionate and methylmalonate are excreted in the urine. Two
types of heritable methylmalonic acidemia and aciduria have been described: one
in which the mutase is missing or deficient (or has reduced affinity for the
coenzyme), and one in which the patient is unable to convert vitamin B12
into its coenzyme form. Either type results in metabolic acidosis and
neurologic manifestations.]
7. Oxidation of unsaturated fatty acids: The oxidation of unsaturated fatty
acids provides less energy than that of saturated fatty acids because
unsaturated fatty acids are less highly reduced, and, therefore, fewer reducing
equivalents can be produced from these structures. Oxidation of monounsaturated
fatty acids, such as 18:1(9) (oleic acid), requires one additional enzyme,
3,2-enoyl CoA isomerase, which converts the 3-cis derivative obtained after
three rounds of β-oxidation to the 2-trans derivative required as a substrate
by the enoyl CoA hydratase. Oxidation of polyunsaturated fatty acids, such as
18:2(9,12) (linoleic acid), requires an NADPH-dependent 2,4-dienoyl CoA
reductase in addition to the isomerase.
8. β-Oxidation in the peroxisome: VLCFAs of more than 22 carbons undergo a preliminary β-oxidation in peroxisomes, because peroxisomes are the primary site of t he synthetase that activates fatty acids of this length. The shortened fatty acid (linked to carnitine) diffuses to a mitochondrion for further oxidation. In contrast to mitochondrial β-oxidation, the initial dehydrogenation in peroxisomes is catalyzed by a FAD-containing acyl CoA oxidase. The FADH2 produced is oxidized by molecular oxygen, which is reduced to H2O2. Therefore, no ATP is generated by this step. The H2O2 is reduced to H2O by catalase. [Note: Genetic defects in the ability either to target matrix proteins to peroxisomes (resulting in Zellweger syndrome, a peroxisomal biogenesis disorder) or to transport VLCFAs across the peroxisomal membrane (resulting in X-linked adrenoleukodystrophy), lead to accumulation of VLCFAs in the blood and tissues.]
C. Peroxisomal α-oxidation of fatty acids
Branched-chain phytanic
acid: This product of chlorophyll metabolism is not a substrate for acyl CoA
dehydrogenase because of the methyl group on its β-carbon (Figure 16.21).
Instead, it is hydroxylated at the α-carbon by phytanoyl CoA α-hydroxylase
(PhyH), carbon 1 is released as CO2, and the product, 19-carbon
pristanal, is oxidized to pristanic acid, which is activated to its CoA
derivative and undergoes β-oxidation. Refsum disease is a rare,
autosomal-recessive disorder caused by a deficiency of peroxisomal PhyH. This
results in the accumulation of phytanic acid in the plasma and tissues. The
symptoms are primarily neurologic, and the treatment involves dietary
restriction to halt disease progression. [Note: ω-Oxidation (at the methyl
terminus) also is known and generates dicarboxylic acids. Normally a minor pathway
of the ER, its upregulation is seen with conditions such as MCAD deficiency
that limit fatty acid β-oxidation.]
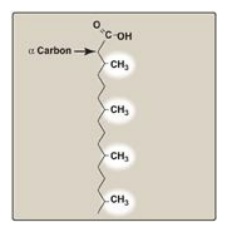
Figure 16.21 Phytanic acid, a
branched-chain fatty acid.
Related Topics
