Chapter Summary, Questions Answers - Fatty Acid, Ketone Body, and Triacylglycerol Metabolism
| Home | | Biochemistry |Chapter: Biochemistry : Fatty Acid, Ketone Body, and Triacylglycerol Metabolism
Generally a linear hydrocarbon chain with a terminal carboxyl group, a fatty acid can be saturated or unsaturated. Two fatty acids are dietary essentials: linoleic and α-linolenic acids.
CHAPTER SUMMARY
Generally a linear
hydrocarbon chain with a terminal carboxyl group, a fatty acid can be saturated
or unsaturated. Two fatty acids are dietary essentials: linoleic and
α-linolenic acids. Fatty acids are synthesized in the cytosol of liver
following a meal containing excess carbohydrate and protein. Carbons used to
synthesize fatty acids are provided by acetyl coenzyme A (CoA), energy by ATP,
and reducing equivalents by nicotinamide adenine dinucleotide phosphate
([NADPH]; Figure 16.25) provided by the pentose phosphate pathway and malic
enzyme. Citrate carries two-carbon acetyl units from the mitochondrial matrix to
the cytosol. The regulated step in fatty acid synthesis is catalyzed by
biotin-requiring acetyl CoA carboxylase (ACC) . Citrate allosterically
activates ACC and long-chain fatty acyl CoAs inhibit it. ACC can also be
activated by insulin and inactivated by adenosine monophosphate–activated
protein kinase (AMPK) in response to epinephrine, glucagon, or a rise in AMP.
The remaining steps in fatty acid synthesis are catalyzed by the
multifunctional enzyme, fatty acid synthase, which produces palmitoyl CoA by
adding two-carbon units from malonyl CoA to a series of acyl acceptors. Fatty
acids can be elongated and desaturated in the endoplasmic reticulum (ER). When
fatty acids are required for energy, adipocyte hormone-sensitive lipase
(activated by epinephrine, and inhibited by insulin), along with other lipases,
degrades stored triacylglycerol (TAG). The fatty acid products are carried by
serum albumin to the liver and peripheral tissues, where oxidation of the fatty
acids provides energy. The glycerol backbone of the degraded TAG is carried by
the blood to the liver, where it serves as an important gluconeogenic
precursor. Fatty acid degradation (β-oxidation) occurs in mitochondria. The
carnitine shuttle is required to transport long-chain fatty acids from the cytosol
to the mitochondrial matrix. A translocase and the enzymes carnitine
palmitoyltransferases (CPT) I and II are required. CPT-I is inhibited by
malonyl CoA, thereby preventing simultaneous synthesis and degradation of fatty
acids. In the mitochondria, fatty acids are oxidized, producing acetyl CoA,
nicotinamide adenine dinucleotide (NADH), and flavin adenine dinucleotide (FADH2).
The first step in the β-oxidation pathway is catalyzed by one of four acyl CoA
dehydrogenases, each with chain-length specificity. Medium-chain fatty acyl CoA
dehydrogenase (MCAD) deficiency causes a decrease in fatty acid oxidation
(process stops once a medium chain fatty acid is produced), resulting in
hypoketonemia and severe hypoglycemia. Oxidation of fatty acids with an odd
number of carbons proceeds two carbons at a time (producing acetyl CoA) until
three-carbon propionyl CoA remains. This compound is carboxylated to
methylmalonyl CoA (by biotin-requiring propionyl CoA carboxylase), which is
then converted to succinyl CoA (a gluconeogenic precursor) by vitamin
B2-requiring methylmalonyl CoA mutase. A genetic error in the mutase or vitamin
B12 deficiency causes methylmalonic acidemia and aciduria.
β-Oxidation of very-long-chain fatty acids and α-oxidation of branched-chain
fatty acids occur in the peroxisome. ω-Oxidation, a minor pathway, occurs in
the ER. Liver mitochondria can convert acetyl CoA derived from fatty acid oxidation
into the ketone bodies acetoacetate and 3-hydroxybutyrate. Peripheral tissues
possessing mitochondria can oxidize 3-hydroxybutyrate to acetoacetate, which
can be reconverted to acetyl CoA, thereby producing energy for the cell. Unlike
fatty acids, ketone bodies are utilized by the brain and, therefore, are
important fuels during a fast. Because the liver lacks the ability to degrade
ketone bodies, it synthesizes them specifically for the peripheral tissues.
Ketoacidosis occurs when the rate of ketone body formation is greater than the
rate of use, as is seen in cases of uncontrolled type 1 diabetes mellitus.
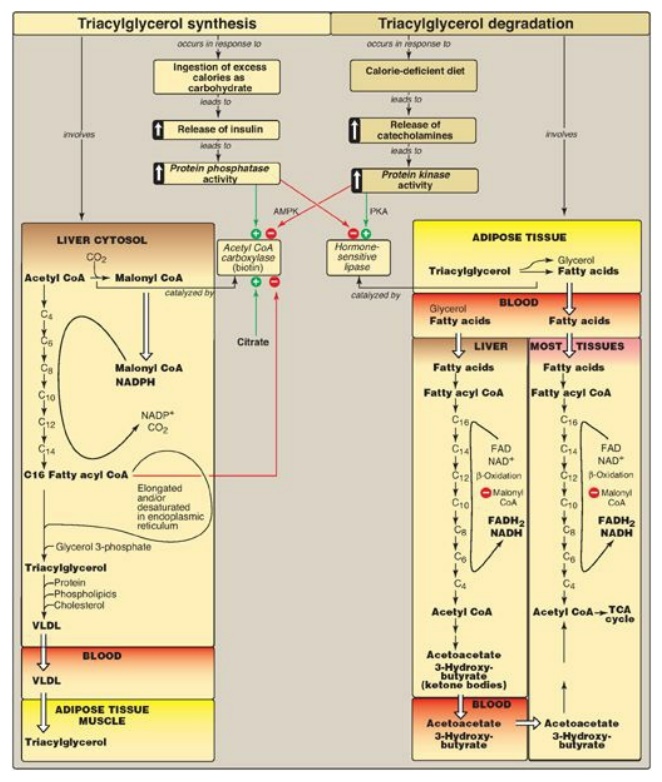
Figure 16.25 Key concept map for fatty acid and triacylglycerol metabolism. AMPK = adenosine monophosphate-activated protein kinase; PKA = protein kinase A; CoA = coenzyme A; NADP(H) = nicotinamide adenine dinucleotide phosphate; FAD(H2) = flavin adenine dinucleotide; NAD(H) = nicotinamide adenine dinucleotide; TCA = tricarboxylic acid; VLDL = very-low-density lipoprotein.
Study Questions
Choose the ONE correct answer.
16.1 When oleic acid, 18:1(9), is desaturated at carbon
6 and then elongated, what is the product?
A. 19:2(7,9)
B. 20:2 (n-6)
C. 20:2(6,9)
D. 20:2(8,11)
Correct answer = D. Fatty acids are elongated in the endoplasmic reticulum by adding two carbons at a time to the carboxylate end (carbon 1) of the molecule. This pushes the double bonds at carbon 6 and carbon 9 further away from carbon 1. 20:2(8,11) is an n-9 (ω-9) fatty acid.
16.2 A 4-month-old child is being evaluated for
fasting hypoglycemia. Laboratory tests at admission reveal low levels of ketone
bodies, free carnitine, and acylcarnitines in the blood. Free fatty acid levels
in the blood were elevated. Deficiency of which of the following would best
explain these findings?
A. Adipose triglyceride
lipase
B. Carnitine transporter
C. Carnitine
palmitoyltransferase I
D. Long-chain fatty
acid dehydrogenase
Correct answer = B. A defect in the carnitine
transporter (primary carnitine deficiency) would result in low levels of
carnitine in the blood (as a result of increased urinary loss) and low levels
in the tissues. In the liver, this decreases fatty acid oxidation and
ketogenesis. Consequently, blood levels of free fatty acids rise. Deficiencies
of adipose triglyceride lipase would decrease fatty acid availability.
Deficiency of carnitine palmitoyltransferase I
would result in elevated blood carnitine. Defects in any of the enzymes of
β-oxidation would result in secondary carnitine deficiency, with a rise in
acylcarnitines.
16.3 A teenager, concerned about his weight,
attempts to maintain a fat-free diet for a period of several weeks. If his
ability to synthesize various lipids were examined, he would be found to be most
deficient in his ability to synthesize:
A. cholesterol.
B. glycolipids.
C. phospholipids.
D. prostaglandins.
E. triacylglycerol.
Correct answer = D. Prostaglandins are synthesized
from arachidonic acid. Arachidonic acid is synthesized from linoleic acid, an
essential fatty acid obtained by humans from dietary lipids. The teenager would
be able to synthesize all other compounds but, presumably, in somewhat
decreased amounts.
16.4 A 6-month-old boy was hospitalized following a
seizure. History revealed that for several days prior, his appetite was
decreased due to a “stomach virus.” At admission, his blood glucose was 24
mg/dl (age-referenced normal is 60–100). His urine was negative for ketone
bodies and positive for a variety of dicarboxylic acids. Blood carnitine levels
were normal. A tentative diagnosis of medium-chain fatty acyl coenzyme A
dehydrogenase (MCAD) deficiency is made. In patients with MCAD deficiency, the
fasting hypoglycemia is a consequence of:
A. decreased acetyl coenzyme A production.
B. decreased ability to
convert acetyl coenzyme A to glucose.
C. increased conversion
of acetyl coenzyme A to acetoacetate.
D. increased production
of ATP and nicotinamide adenine dinucleotide.
Correct answer = A. Impaired oxidation of fatty acids
less than 12 carbons in length results in decreased production of acetyl
coenzyme (CoA), the allosteric activator of pyruvate carboxylase, a
gluconeogenic enzyme, and, thus, glucose levels fall. Acetyl CoA can never be
used for the net synthesis of glucose. Acetoacetate is a ketone body, and with
medium-chain fatty acyl CoA dehydrogenase deficiency, ketogenesis is decreased
as a result of decreased production of the substrate, acetyl
CoA. Impaired fatty
acid oxidation means that less ATP and nicotinamide adenine dinucleotide are
made, and both are needed for gluconeogenesis.
16.5 Explain why with Zellweger syndrome both
very-long-chain fatty acids (VLCFAs) and phytanic acid accumulate, whereas with
X-linked adrenoleukodystrophy, only VLCFAs accumulate.
Zellweger syndrome is
caused by an inability to target matrix proteins to the peroxisome. Therefore,
all peroxisomal activities are affected because functional peroxisomes are not
able to be formed. In X-linked adrenoleukodystrophy, the defect is an inability
to transport very-long-chain fatty acids into the peroxisome, but other
peroxisomal functions, such as α-oxidation, are normal.
Related Topics
