Synthesis of Glycoproteins
| Home | | Biochemistry |Chapter: Biochemistry : Glycosaminoglycans, Proteoglycans, and Glycoproteins
Proteins destined to function in the cytoplasm are synthesized on free cytosolic ribosomes.
SYNTHESIS OF GLYCOPROTEINS
Proteins destined to
function in the cytoplasm are synthesized on free cytosolic ribosomes. However,
proteins, including glycoproteins, that are destined for cellular membranes,
lysosomes, or to be exported from the cell, are synthesized on ribosomes
attached to the RER. These proteins contain specific signal sequences that act
as molecular “address labels,” targeting the proteins to their proper
destinations. An N-terminal hydrophobic sequence initially directs these
proteins to the RER, allowing the growing polypeptide to be extruded into the
lumen. The proteins are then transported via secretory vesicles to the Golgi
complex, which acts as a sorting center (Figure 14.15). In the Golgi, those
glycoproteins that are to be secreted from the cell (or are targeted for
lysosomes) are packaged into vesicles that fuse with the cell (or lysosomal)
membrane and release their contents. Those that are destined to become
components of the cell membrane are integrated into the Golgi membrane, which
buds off, forming vesicles that add their membrane-bound glycoproteins to the
cell membrane. [Note: The membrane glycoproteins are, thus, oriented with the
carbohydrate portion on the outside of the cell (see Figure 14.15).]
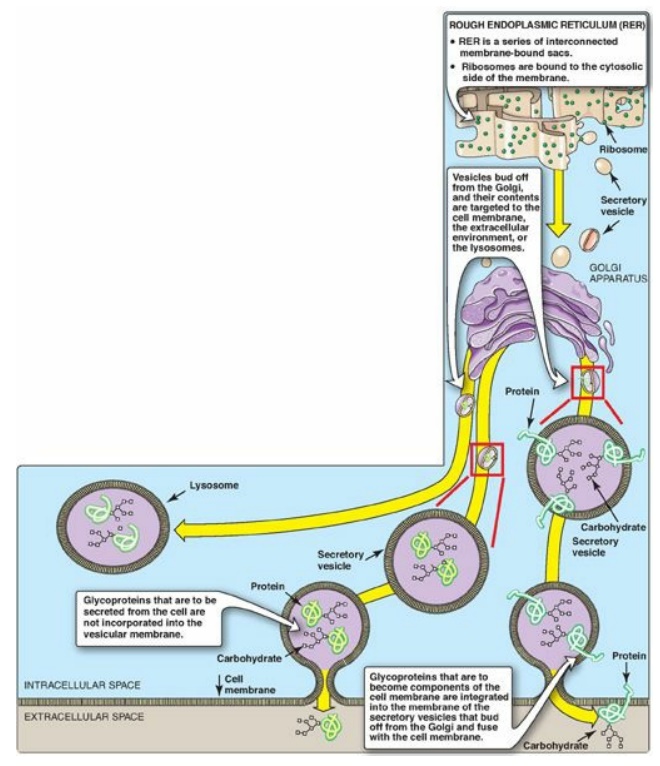
Figure 14.15 Transport of glycoproteins through the Golgi apparatus and their subsequent release or incorporation into a lysosome or the cell membrane.
A. Carbohydrate components of glycoproteins
The precursors of the
carbohydrate components of glycoproteins are nucleotide sugars, which include UDP-glucose,
UDP-galactose, UDP-GlcNAc, and UDP-GalNAc. In addition, guanosine diphosphate
(GDP)-mannose, GDP-L-fucose (which is synthesized from GDP-mannose), and
CMP-NANA may donate sugars to the growing chain. [Note: When the acidic NANA is
present, the oligosaccharide has a negative charge at physiologic pH.] The
oligosaccharides are covalently attached to the R groups of specific amino
acids in the protein, where the three-dimensional structure of the protein
determines whether or not a specific amino acid is glycosylated.
B. Synthesis of O-linked glycosides
The synthesis of the
O-linked glycosides is very similar to that of the GAGs. First, the protein to
which the oligosaccharides are to be attached is synthesized on the RER and
extruded into its lumen. Glycosylation begins with the transfer of GalNAc (from
UDP-GalNAc) onto the R-group of a specific serine or threonine.
1. Role of glycosyltransferases: The glycosyltransferases
responsible for the stepwise synthesis of the oligosaccharides are bound to the
membranes of the Golgi apparatus. They act in a specific order, without using a
template as is required for DNA, RNA, and protein synthesis (see Unit VI) but,
rather by recognizing the actual structure of the growing oligosaccharide as
the appropriate substrate.
C. Synthesis of the N-linked glycosides
The synthesis of N-linked glycosides occurs in the lumen of the RER and requires the participation of the phosphorylated form of dolichol (dolichol pyrophosphate), a lipid of the ER membrane (Figure 14.16). The initial product is processed in the ER and Golgi.
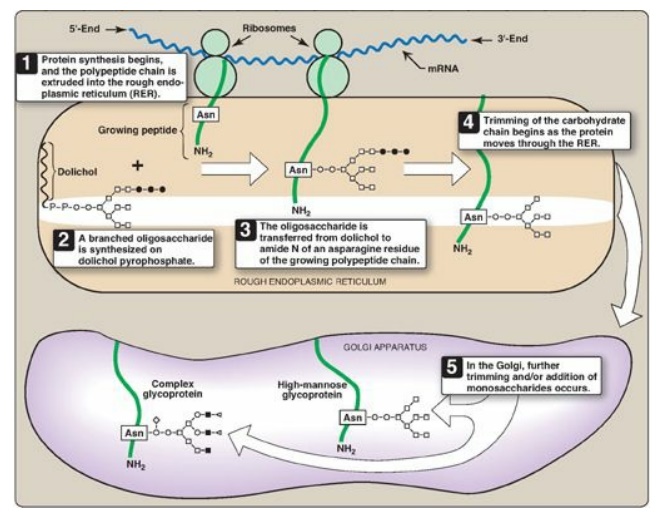
Figure 14.16 Synthesis of N-linked glycoproteins. o= N-acetylglucosamine; □= mannose; ●= glucose; ■ = N-acetylgalactosamine; ◄ or ◊ = terminal group (fucose or N-acetylneuraminic acid); mRNA = messenger RNA; Asn = asparagine.
1. Synthesis of dolichol-linked oligosaccharide: First, as with the O-linked
glycosides, the protein is synthesized on the RER and enters its lumen.
However, the protein does not become glycosylated with individual sugars.
Instead, a lipid-linked oligosaccharide is first constructed. This consists of
dolichol (an ER membrane lipid 80–100 carbons long) attached through a
pyrophosphate linkage to an oligosaccharide containing N-GlcNAc, mannose, and glucose.
The sugars to be added sequentially to the dolichol by the membrane-bound
glycosyltransferases are first N-GlcNAc, followed by mannose and glucose (see
Figure 14.16). The entire 14-sugar oligosaccharide is then transferred from the
dolichol to the amide nitrogen (N) of an asparagine in the protein to be
glycosylated by a protein-oligosaccharide transferase present in the ER. [Note:
Tunicamycin inhibits N-linked glycosylation.]
Congenital disorders of glycosylation (CDGs) are syndromes caused primarily by defects in the N-linked glycosylation of proteins, either oligosaccharide assembly (Type I) or processing (Type II).
2. Final processing of N-linked oligosaccharides: After incorporation into the protein, the N-linked oligosaccharide is processed by the removal of specific mannosyl and glucosyl residues as the glycoprotein moves through the RER. Finally, the oligosaccharide chains are completed in the Golgi by addition of a variety of sugars (for example, N-GlcNAc, N-GalNAc, and additional mannoses, and then fucose or NANA as terminal groups) to produce a complex glycoprotein. Alternatively, they are not processed further, leaving branched, mannose-containing chains in a high-mannose glycoprotein (see Figure 14.16). The ultimate fate of N-linked glycoproteins is the same as that of the O-glycoproteins linked (for example, they can be released by the cell or become part of a cell membrane). In addition, N-linked glycoproteins can be targeted to the lysosomes. [Note: Nonenzymatic glycosylation of proteins is known as glycation.]
3. Enzymes destined for
lysosomes: N-linked glycoproteins being processed through the Golgi can be
phosphorylated on carbon 6 of one or more specific mannosyl residues. Mannose
6-phosphate receptors, located in the Golgi apparatus, bind the mannose
6-phosphate residues of these targeted enzymes, which are then packaged into
vesicles and sent to the lysosomes. I-cell disease is a rare lysosomal storage
disease in which the acid hydrolases normally found in lysosomes are absent,
resulting in an accumulation of substrates normally degraded by these enzymes.
[Note: I-cell disease is so-named because of the large inclusion bodies seen in
cells of patients with this disease.] In addition, high amounts of lysosomal
enzymes are found in the patient’s plasma and urine, indicating that the
targeting process to lysosomes (rather than the synthetic pathway of these
enzymes) is deficient. Individuals with I-cell disease are lacking the
phosphotransferase needed to phosphorylate the mannose residues of potential
lysosomal enzymes, causing the enzymes to be secreted (by default), rather than
being targeted to lysosomal vesicles (Figure 14.17). I-cell disease is
characterized by skeletal abnormalities, restricted joint movement, coarse
(dysmorphic) facial features, and severe psychomotor impairment. [Note: Because
I-cell disease has features in common with the mucopolysaccharidoses and
sphingolipidoses, it is termed a mucolipidosis.] Currently, there is no cure,
and death from cardiopulmonary complications usually occurs by age 10 years.
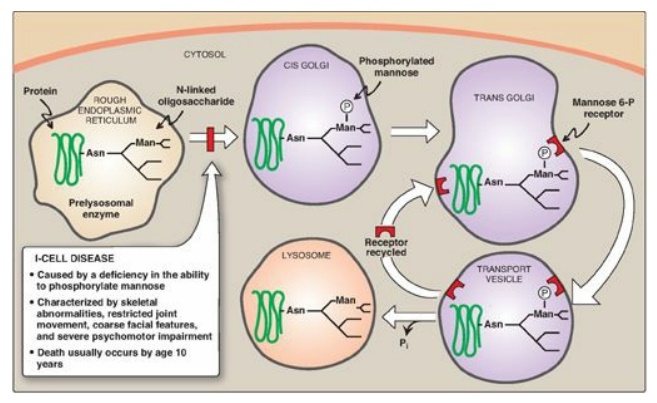
Figure 14.17 Mechanism for transport of N-linked glycoproteins to the lysosomes. Asn = asparagine; Man = mannose; P = phosphate; Pi = inorganic phosphate.
Related Topics
