Degradation of Purine Nucleotides
| Home | | Biochemistry |Chapter: Biochemistry : Nucleotide Metabolism
Degradation of dietary nucleic acids occurs in the small intestine, where a family of pancreatic enzymes hydrolyzes the nucleic acids to nucleotides.
DEGRADATION OF PURINE NUCLEOTIDES
Degradation of dietary
nucleic acids occurs in the small intestine, where a family of pancreatic
enzymes hydrolyzes the nucleic acids to nucleotides. Inside the intestinal
mucosal cells, purine nucleotides are sequentially degraded by specific enzymes
to nucleosides and free bases, with uric acid as the end product of this
pathway. [Note: Purine nucleotides from de novo synthesis are degraded in the
liver primarily. The free bases are sent out from liver and salvaged by
peripheral tissues.]
A. Degradation of dietary nucleic acids in the small intestine
Ribonucleases and
deoxyribonucleases, secreted by the pancreas, hydrolyze dietary RNA and DNA to
oligonucleotides. Oligonucleotides are further hydrolyzed by pancreatic
phosphodiesterases, producing a mixture of 3 I - and 5 I -mononucleotides.
In the intestinal mucosal cells, a family of nucleotidases removes the
phosphate groups hydrolytically, releasing nucleosides that are further
degraded by nucleosidases (nucleoside phosphorylases) to free bases plus
(deoxy) ribose 1-phosphate. Dietary purine bases are not used to any
appreciable extent for the synthesis of tissue nucleic acids. Instead, they are
generally converted to uric acid in intestinal mucosal cells. Most of the uric
acid enters the blood and is eventually excreted in the urine. A summary of
this pathway is shown in Figure 22.14. [Note: Mammals other than primates
express urate oxidase (uricase) which cleaves the purine ring, generating
allantoin. Modified recombinant urate oxidase is now used clinically to lower
urate levels.]
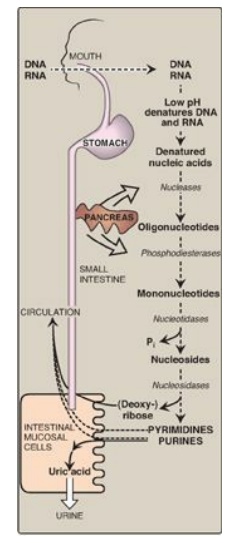
Figure 22.14 Digestion of dietary nucleic acids. [Note: Much of the metabolism of the mononucleotides occurs within the intestinal mucosal cells.] Pi = inorganic phosphate.
B. Formation of uric acid
A summary of the steps
in the production of uric acid and genetic diseases associated with
deficiencies of specific degradative enzymes are shown in Figure 22.15. [Note:
The bracketed numbers refer to specific reactions in the figure.]
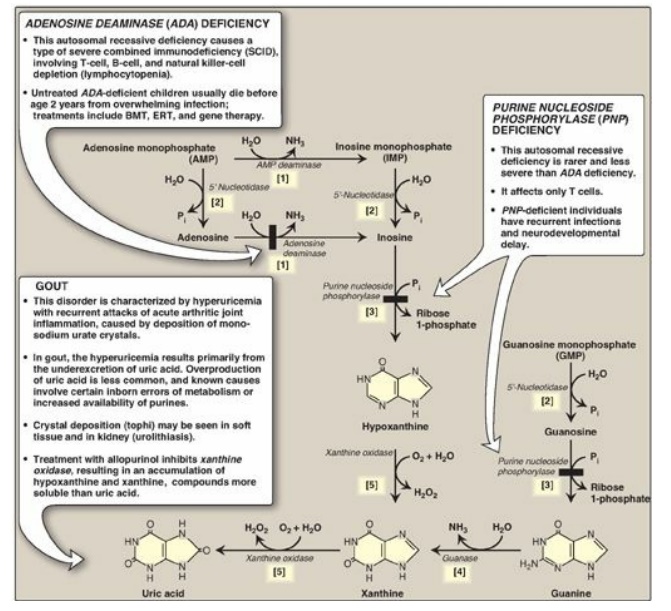
Figure 22.15 The degradation of purine nucleotides to uric acid, illustrating some of the genetic diseases associated with this pathway. [Note: The numbers in brackets refer to the corresponding numbered citations in the text.] BMT = bone marrow transplantation; ERT = enzyme replacement therapy; Pi = inorganic phosphate.
[1] An amino group is
removed from AMP to produce IMP by AMP deaminase or from adenosine to produce
inosine (hypoxanthine-ribose) by adenosine deaminase.
[2] IMP and GMP are
converted into their nucleoside forms (inosine and guanosine) by the action of
5 -nucleotidase.
[ 3 ] Purine nucleoside
phosphorylase converts inosine and guanosine into their respective purine
bases, hypoxanthine and guanine. [Note: A mutase interconverts ribose 1- and
ribose 5-phosphate.]
[4] Guanine is
deaminated to form xanthine.
[5] Hypoxanthine is
oxidized by xanthine oxidase to xanthine, which is further oxidized by xanthine
oxidase to uric acid, the final product of human purine degradation. Uric acid
is excreted primarily in the urine.
C. Diseases associated with purine degradation
Gout: Gout is a disorder initiated by high
levels of uric acid (the end product of purine catabolism) in blood
(hyperuricemia), as a result of either the overproduction or underexcretion of
uric acid. The hyperuricemia can lead to the deposition of monosodium urate (MSU)
crystals in the joints and an inflammatory response to the crystals, causing
first acute and then progressing to chronic gouty arthritis. Nodular masses of
MSU crystals (tophi) may be deposited in the soft tissues, resulting in chronic
tophaceous gout (Figure 22.16). Formation of uric acid stones in the kidney
(urolithiasis) may also be seen. [Note: Hyperuricemia, while necessary, is not
sufficient to cause gout, but gout is always preceded by hyperuricemia.
Hyperuricemia is typically asymptomatic but may be indicative of comorbid
conditions such as hypertension.] The definitive diagnosis of gout requires
aspiration and examination of synovial fluid (Figure 22.17) from an affected
joint (or material from a tophus) using polarized light microscopy to confirm
the presence of needle-shaped MSU crystals (Figure 22.18).
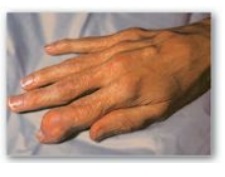
Figure 22.16 Tophaceous gout.
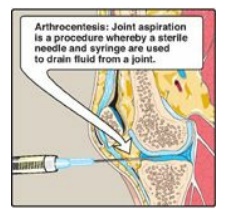
Figure 22.17 Analysis of joint fluid can help to define causes of joint swelling or arthritis, such as infection, gout, and rheumatoid disease.
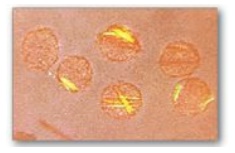
Figure 22.18 Gout can be diagnosed by the presence of negatively birefringent monosodium urate crystals in aspirated synovial fluid examined by polarized-light microscopy. Here, crystals are within polymorphonuclear leukocytes.
a. Underexcretion of uric acid: In over 90% of individuals, hyperuricemia is caused by underexcretion of uric acid. Underexcretion can be primary, due to as-yet-unidentified inherent excretory defects, or secondary to known disease processes that affect how the kidney handles urate (for example, in lactic acidosis, lactate increases renal urate reabsorption, thereby decreasing its excretion) and to environmental factors such as the use of drugs (for example, thiazide diuretics) or exposure to lead (saturnine gout).
b. Overproduction of uric acid: A less common cause of hyperuricemia is from the overproduction of uric acid. Primary hyperuricemia is, for the most part, idiopathic (having no known cause). However, several identified mutations in the gene for X-linked PRPP synthetase result in the enzyme having an increased maximal velocity (Vmax) for the production of PRPP, a lower Km for ribose 5-phosphate, or a decreased sensitivity to purine nucleotides, its allosteric inhibitors. In each case, increased availability of PRPP increases purine production, resulting in elevated levels of plasma uric acid. Lesch-Nyhan syndrome also causes hyperuricemia as a result of the decreased salvage of hypoxanthine and guanine and the subsequent increased availability of PRPP. Secondary hyperuricemia is typically the consequence of increased availability of purines (for example, in patients with myeloproliferative disorders or who are undergoing chemotherapy and so have a high rate of cell turnover). Hyperuricemia can also be the result of seemingly unrelated metabolic diseases, such as von Gierke disease or hereditary fructose intolerance.
A diet rich in meat, seafood (particularly
shellfish), and ethanol is associated with increased risk of gout, whereas a
diet rich in low-fat dairy products is associated with a decreased risk.
c. Treatment of gout: Acute attacks of gout are treated with anti-inflammatory agents. Colchicine; steroidal drugs, such as prednisone; and nonsteroidal drugs, such as indomethacin, are used. [Note: Colchicine prevents formation of microtubules, thereby decreasing the movement of neutrophils into the affected area. Like the other anti-inflammatory drugs, it has no effect on uric acid levels.] Long-term therapeutic strategies for gout involve lowering the uric acid level below its saturation point (6.5 mg/dl), thereby preventing the deposition of urate crystals. Uricosuric agents, such as probenecid or sulfinpyrazone, that increase renal excretion of uric acid, are used in patients who are “underexcretors” of uric acid. Allopurinol, a structural analog of hypoxanthine, inhibits uric acid synthesis and is used in patients who are “overproducers” of uric acid. Allopurinol is converted in the body to oxypurinol, which inhibits xanthine oxidase (XO), resulting in an accumulation of hypoxanthine and xanthine (see Figure 22.15), compounds more soluble than uric acid and, therefore, less likely to initiate an inflammatory response. In patients with normal levels of HGPRT, the hypoxanthine can be salvaged, reducing the levels of PRPP and, therefore, de novo purine synthesis. Febuxostat, a non-purine inhibitor of XO, is also available. [Note: Uric acid levels in the blood normally are close to the saturation point. One reason for this may be the strong antioxidant effects of uric acid.]
2. Adenosine deaminase deficiency: Adenosine deaminase (ADA) is expressed in a variety of tissues, but, in humans, lymphocytes have the highest activity of this cytoplasmic enzyme. A deficiency of ADA results in an accumulation of adenosine, which is converted to its ribonucleotide or deoxyribonucleotide forms by cellular kinases. As dATP levels rise, ribonucleotide reductase is inhibited, thereby preventing the production of all deoxyribose-containing nucleotides. Consequently, cells cannot make DNA and divide. [Note: The dATP and adenosine that accumulate in ADA deficiency lead to developmental arrest and apoptosis of lymphocytes.] In its most severe form, this autosomal-recessive disorder causes a type of severe combined immunodeficiency disease (SCID), involving a decrease in T cells, B cells, and natural killer cells. It is estimated that, in the United States, ADA deficiency accounts for approximately 14% of all cases of SCID. Treatments include bone marrow transplantation, enzyme replacement therapy, and gene therapy. Without appropriate treatment, children with this disorder usually die from infection by age 2 years. [Note: Purine nucleoside phosphorylase deficiency results in a less severe immunodeficiency primarily involving T cells.]
Related Topics
